
Jeffrey Kephart, Steve White
Proceedings of the IEEE Computer Society Symposium on Research in Security and Privacy, pp. 343-359
1991
 Download PDF (1.59Mb) (You need to be registered on forum)
Download PDF (1.59Mb) (You need to be registered on forum)Despite serious concerns raised by the proven ability of computer viruses to spread between individual systems and establish themselves as a persistent infection in the computer population, there have been very few efforts to analyze their propagation theoretically. The strong analogy between biological viruses and their computational counterparts has motivated us to adapt the techniques of mathematical epidemiology to the study of computer-virus propagation. In order to allow for the most general patterns of program sharing, we extend a standard epidemiological model by placing it on a directed graph and use a combination of analysis and simulation to study its behavior. We determine the conditions under which epidemics are likely to occur, and in cases where they do, we explore the dynamics of the expected number of infected individuals as a function of time. We conclude that an imperfect defense against computer viruses can still be highly effective in preventing their widespread proliferation, provided that the infection rate does not exceed a well-defined critical epidemic threshold.
A fascination with the potential ability of computer programs to mimic biological processes such as self-replication and the workings of the human brain dates from the earliest days of computer science [1, 2, 3]. Until recently, much of the work in this field (with the notable exception of genetic algorithms [4]) was relegated to sporadic reporting in the Mathematical Games department in Scientific American. In the last few years, however, a remarkable resurgence of interest in the analogies between biological and computational mechanisms has revitalized a number of long-neglected fields, including neural networks [5], cellular automata [6], and artificial life [7]. Recently, a new concept has added itself to this intellectual milieu: the computer virus [8] - a self-replicating program that spreads within computing systems, either by attaching itself parasitically to existing programs or by spawning self-sufficient copies of itself1. The compelling analogies that exist between computer viruses and their biological counterparts have been apparent ever since the term was coined [8]. This has led some authors to suggest that the mathematical techniques which have been developed for the study of the spread of infectious diseases might be adapted to the study of the spread of computer viruses [8, 9]. We believe that. this paper represents the first serious attempt to adapt matthematical epidemiology to this problem.
In the remainder of this section, we give a very brief history of the problem of computer viruses and describe our objectives in stiidying them. Then, we critique some previous work on the spread of computer viruses, following it with a similar discussion of the relevant literature on mathematical epidemiology2. We conclude that the standard epidemiological approach has much to offer, but that it must be augmented in order to account properly for the localized nature of program sharing (one of the major vectors for viral infection). Finally, we propose a model which explicitly incorporates such locality, discuss our approach to studying the behavior of that, model, and provide an outline for the remainder of the paper.
Computer viruses were originally thought of as problematic primarily because of their ability to carry out directed attacks against isolated systems [8]. Their potential ability to flow from user to user in a system meant that attacks could reach parts of the system that had been thought to be more secure. In the last few years, however, as viruses have been written and released outside of controlled environments and into the world’s computing community, their ability to spread between individual systems and thus affect a global collection of systems has proved to be of greater concern.
Since the first documented reports of microcomputer viruses in the mid-1980’s [10, 11], they have spread throughout the world. At this date, we estimate very conservatively that there are many thousands of microcomputers with active virus infections, and this population of infected systems continues to spread the infection. The Internet Worm [12, 13, 14, 15] infected at least hundreds and probably thousands of computers on the Internet in the space of a few hours in November, 1988, with the resultant loss of a substantial amount of service and many hours devoted to expurgating it from systems.
Our long-term goal is to develop and analyze quantitatively models which capture the spreading characteristics of computer viruses. The potential benefits of doing so can be divided into two major categories.
First, mathematical models could aid in the evaluation and development of general policies and heuristics for inhibiting the spread of viruses. Although it is well known that a general-purpose computing system need only satisfy minimal conditions to be capable in principle of being completely infiltrated by a virus [8],the set of conditions under which this is likely to occur may be considerably more restricted. We wish to gain a quantitative understanding of the vulnerability of current systems to viral infections and to determine the effectiveness of proposed heuristics for inhibiting viral spread [16, 17, 18, 19, 20]. In a similar vein, mathematical modelling of the sort we describe could be helpful in the design of new systems - allowing a reasonable tradeoff between the ease with which legitimate programs can flow and the ease with which viruses can spread.
A second major use for mathematical modelling, more in the spirit of biological epidemiology, is to apply it to a particular epidemic. In its more passive application, modelling can aid in predicting the course of a particular epidemic, so as to plan what resources will be needed to deal with the problem. A more aggressive role for modelling, which is gaining popularity in the biological realm, is to use it to determine the optimal policy for controlling the course of a particular epidemic by isolating or immunizing the population at appropriate times [24].
In this paper, we shall deduce a number of general properties of the spread of computer viruses from simple models which capture some essential features of the networks in which they propagate. We believe that this work is an important first step towards a theory which ultimately will be sufficiently realistic to evaluate specific proposals for thwarting the spread of computer viruses.
Cohen was the first to define and describe computer viruses in their present form [8]. He demonstrated that, in the worst case, infection can spread to the transitive closure of information flow in a system. In other words, if A can infect B and B can infect C, a virus that originates with A can propagate to C. He performed extensive experiments on a variety of systems (most of them multi-user) which demonstrated that a virus could propagate to a level of security higher than that from which it had originated. He and Murray [9] pointed out the connection between computer virus spread and biological epidemiology, but neither pursued it to the point of developing an explicit model.
Recently, there have been attempts to describe viral spread more quantitatively. Gleissner [21] examined a model of computer virus spread on a multi-user system. Quantitative analysis of the model reproduced Cohen’s result that a virus would reach the transitive closure of information flow, and showed that this could occur at an exponential rate. However, the usefulness of these results was limited because no allowance was made for the fact that individual users of the system might detect and remove viruses or alert other users to their presence. Tippett [22] used the well-known fact that many population models exhibit exponential growth in their initial phases to suggest that the computer virus population might grow to worrisome proportions. However, he did not justify the application of such models to the spread of computer viruses, and the paucity of data on the actual spread of computer viruses makes any such extrapolation extremely suspect. Jones and White [23] examined an analogy between viral spread and infestations of crops by insects and other pests, but did not present an explicit model. Their claim that segregating computing resources leads to an increase in the virus pop ulation seems particularly questionable. Solomon [25] studied a deterministic model of computer virus propagation based upon mathematical epidemiology. The quantitative results that he obtained are equivalent to Eq. 2 of this work. He also introduced and analyzed a novel and potentially important form of inter-virus interaction, whereby the increased vigilance of a user who detects any virus will increase his or her probability of detecting other viruses in the future.
The first application of mathematical modelling to the spread of infectious disease was carried out by Daniel Bernoulli in 1760 [26]. Although his work predated the identification of the agent responsible for the transmission of smallpox by a century, he formulated and solved a differential equation describing the dynamics of the infection which is still of value in our day. The development of mathematical epidemiology was stalled by a lack of understanding of the mechanism of infectious spread until the beginning of this century [27]. McKendrick developed the first stochastic theory in 1926 [28], and in the 1930’s Kermack and McKendrick [27] established the extremely important threshold theorem, showing that the density of susceptible individuals must exceed a certain critical value in order for an epidemic to occur. In 1975, Bailey [27] reported that the number of references to mathematical epidemiology had quintupled to 500 in a space of 18 years. Currently, there are several papers on mathematical epidemiology per month in Mathematical Biosciences, one of many journals which publishes such work.
In order to apply this vast catalog of mathematical techniques to the study of computer virus spread, we view a single computing system as an individual in a population of similar individuals. Following the usual epidemiological approach, we neglect the details of infection inside a single system and consider an individual to be in one of a small number of discrete states, e.g. infected, uninfected, immune, etc. One might object to such a simplistic classification because some types of computer viruses, such as those that infect a large class of executable files in a system, can cause a system to become “more” infected over time - thereby increasing the rate at which infection can be transmitted from that system. However, the time scale on which the internal infection occurs is generally much shorter than that on which the infection spreads to other systems, so such a simplification is quite reasonable.
A further simplification which is characteristic of epidemiology is to abstract the details of viral transmission into a probability per unit time that a particular infected individual will infect a particular uninfected individual. Likewise, we abstract the details of detection and removal of a virus into a probability per unit time for an infected individual to be "cured". One could in principle derive the infection rates from the known details of the transmission process and the pattern of program sharing. If this information is unavailable (as it was for Bernoulli), it is often possible to simply measure the rates or infer them by fitting the observed course of an epidemic to a model.
Most current epidemiological models are homogeneous, in the sense that an infected individual is equally likely to infect any of the susceptible individuals. Taken literally, this means that a man sneezing in Chicago is as likely to infect someone in New Delhi as he is someone else in Chicago. This approximation turns out to be adequate for diseases such as influenza, in which the disease can be transmitted via casual contact. However, its validity is generally conceded to be questionable for diseases in which each individual has a limited number of potentially infectious contacts.
Program sharing is far from homogeneous, as one can readily establish by a bit of introspection. Most individuals exchange the majority of their programs with just a few other individuals, and never have any contact with the vast majority of the world's population. Another aspect of program sharing which must be taken into account in models is the fact that it can be strongly asymmetric. For example, the rate at which a retailer ships software greatly exceeds the rate at which a customer sends software to the retailer. Such asymmetry is occasionally important in the biological realm as well, particularly in the case of sexually transmitted diseases.
Recognized deficiencies of the assumption of homogeneous, symmetric interactions have encouraged a variety of attempts to incorporate heterogeneity and asymmetry into biological models. The spatial model is one method that has been used to account for local, symmetric interactions [27]. Local, asymmetric interactions have typically been studied by segregating the population by age, sex, or geographic location, and then treating interactions within the individual subpopulations as homogeneous and symmetric [29, 30, 31]. The model presented in this paper is general enough to encompass both of these approaches, the original homogeneous model, and a variety of other heterogeneous interaction models, some of which we shall explore in this paper.
We account for the heterogeneous communication pattern among individual computer systems in a new and general way: by representing an individual system (a microcomputer, for instance) as a node in a graph. Directed edges from a given node j to other nodes represent the set of individuals that can be infected by j. A rate of infection is associated with each edge. Similarly, a rate at which infection can be detected and "cured" is associated with each node.
Throughout this paper, we shall study one of the simplest of the standard epidemiological models, the SIS (susceptible → infected → susceptible) model, on these graphs. In the SIS model, individuals immediately become susceptible once they are cured of an infection. In our case, this represents an extreme in which users do not become more vigilant after having experienced a viral infection. Our emphasis will be on determining the probability that an infection becomes extinct in a specified population. Under the conditions in which an epidemic is viable, we characterize the expected number of infections as a function of time, particularly equilibria and fluctuations about them. We shall recover some of the well-known results for homogeneous interactions as limiting cases of our more general results.
In the next section, we discuss this model on random graphs. In doing so, we gain a good deal of insight into the relationship between homogeneous and graph models of epidemics and develop a number of useful analytical techniques and approximations. Then, in sections 3 and 4, we investigate the model on hierarchical graphs and N-dimensional Cartesian lattices. We shall conclude in section 5 with a summary of our findings, their potential implications for hindering the spread of computer viruses, and recommendations for future work in this area.
Due to its structural simplicity and the relative ease with which it can be analyzed, the first type of graph on which we shall study virus propagation is a random graph, illustrated in Fig. 1. We construct a random graph of N nodes by making random, independent decisions about whether to include each of the N(N - 1) possible directional edges which can connect two nodes. If p is the probability for a particular edge to be included in the graph, the expected total number of edges is pN(N - 1).
In our version of the SIS model, each edge has associated with it an infection rate 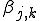 (also referred to as the birth rate of the virus) at which an infected node j can infect an uninfected neighbor k. Similarly, each node j has a cure rate
(also referred to as the birth rate of the virus) at which an infected node j can infect an uninfected neighbor k. Similarly, each node j has a cure rate 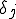 , (or death rate of the virus) at which it will become cured if it is infected. The probabilities per unit time of infection along any edge and of cure of any node are independent. Once an individual is cured, it is immediately capable of being reinfected. The standard homogeneous interaction version of the SIS model is easily recovered from this more general model by connecting all possible pairs of nodes and making all infection and cure rates identical.
, (or death rate of the virus) at which it will become cured if it is infected. The probabilities per unit time of infection along any edge and of cure of any node are independent. Once an individual is cured, it is immediately capable of being reinfected. The standard homogeneous interaction version of the SIS model is easily recovered from this more general model by connecting all possible pairs of nodes and making all infection and cure rates identical.
Our goal is to study the behavior of the SIS model on a typical member of the class of random graphs with N nodes and edge probability p. In the remainder of this section we explore several different techniques for doing so: the deterministic approximation, approximate probabilistic analysis, and simulation.
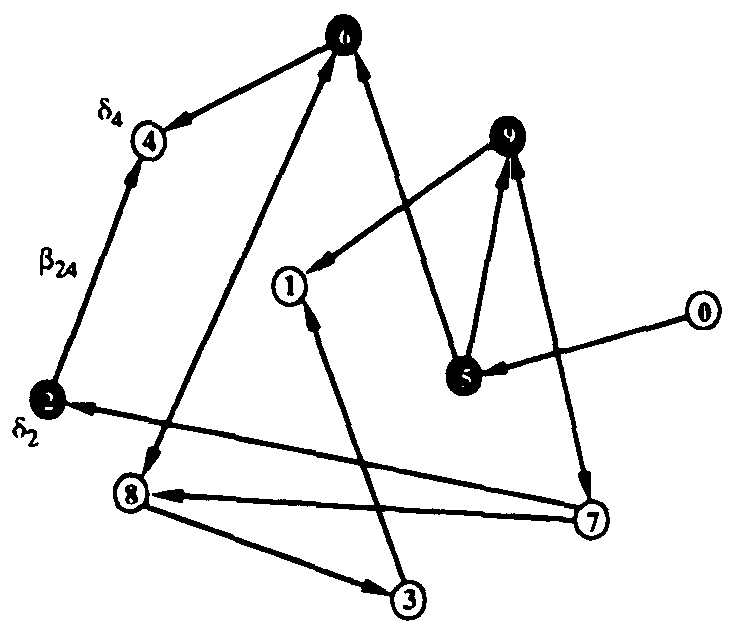
Figure 1: Random graph with 10 nodes. Black filled and unfilled circles represent infected and uninfected nodes, respectively. The probability per unit time that node 2 will infect node 4 is 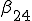 , and the probability per unit time that node 2 will be cured is
, and the probability per unit time that node 2 will be cured is 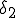 . If node 4 becomes infected by either node 2 or node 6, its probability per unit time of being cured will be
. If node 4 becomes infected by either node 2 or node 6, its probability per unit time of being cured will be 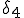 .
.
It is often the case that a deterministic analysis can provide a reasonably accurate picture of many aspects of the dynamics of an epidemic [27], [32]. For the sake of simplicity, we shall assume that the infection rate along each edge is and the cure rate for each node is 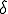 . If the population N is sufficiently large, we can convert I(t), the number of infected nodes in the population at time t, to
. If the population N is sufficiently large, we can convert I(t), the number of infected nodes in the population at time t, to 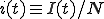 , a continuous quantity representing the fraction of infected nodes. Then, if we assume that the details of the graph’s connectivity are fairly unimportant, the dynamics of infection depend only upon how many nodes are infected (rather than which particular nodes are infected). Later, in section 2.3, when we use simulations to test the validity of this assumption, we shall find that it works well when there are many edges in the graph but fails miserably when there are only a few edges per node.
, a continuous quantity representing the fraction of infected nodes. Then, if we assume that the details of the graph’s connectivity are fairly unimportant, the dynamics of infection depend only upon how many nodes are infected (rather than which particular nodes are infected). Later, in section 2.3, when we use simulations to test the validity of this assumption, we shall find that it works well when there are many edges in the graph but fails miserably when there are only a few edges per node.
Now consider a particular infected node. Since the expected number of edges in the graph is pN(N - 1), the expected number of edges emanating from this node (which we shall refer to as the connectivity3, is 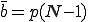 . The fraction of neighbors that are susceptible to infection is 1 - i, so the expected number of uninfected nodes which can be infected by this node is
. The fraction of neighbors that are susceptible to infection is 1 - i, so the expected number of uninfected nodes which can be infected by this node is 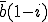 . Therefore, on average we expect the total system-wide rate at which infected nodes infect uninfected nodes to be
. Therefore, on average we expect the total system-wide rate at which infected nodes infect uninfected nodes to be 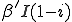 , where
, where 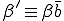 is the average total rate at which a node attempts to infect its neighbors. The system-wide rate at which infected nodes are cured is simply
is the average total rate at which a node attempts to infect its neighbors. The system-wide rate at which infected nodes are cured is simply 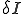 . By ignoring stochastic variation in the number of branches emanating from each node and in the average infection and cure rates, we obtain a deterministic differential equation describing the time evolution of i(t):
. By ignoring stochastic variation in the number of branches emanating from each node and in the average infection and cure rates, we obtain a deterministic differential equation describing the time evolution of i(t):
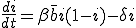 . (1)
. (1)
The solution to Eq. 1 is:
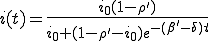 , (2)
, (2)
where 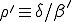 is the average ratio of the rate at which an infected node is cured to that at which it infects other nodes, and
is the average ratio of the rate at which an infected node is cured to that at which it infects other nodes, and 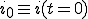 is the initial fraction of infected nodes.
is the initial fraction of infected nodes.
If 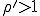 , the fraction of infected individuals decays exponentially from the initial value
, the fraction of infected individuals decays exponentially from the initial value 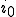 to 0, i.r., there is no epidemic. If
to 0, i.r., there is no epidemic. If 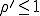 , the fraction of infected individuals grows from the initial value at a rate which is initially exponential (
, the fraction of infected individuals grows from the initial value at a rate which is initially exponential (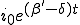 )and eventually saturates at the value
)and eventually saturates at the value 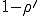 . This result has a simple intuitive interpretation: if the average number of neighbors that an individual can infect during the time that it is infected exceeds one, there will be an epidemic; if this number is less than one, the infection will die out. The existence of this threshold was first established for homogeneous interactions by Kermack and McKendrick [27] in the 1930’s, and here we will show that it holds for directed graphs as well. According to this deterministic result, all that matters is the total rate
. This result has a simple intuitive interpretation: if the average number of neighbors that an individual can infect during the time that it is infected exceeds one, there will be an epidemic; if this number is less than one, the infection will die out. The existence of this threshold was first established for homogeneous interactions by Kermack and McKendrick [27] in the 1930’s, and here we will show that it holds for directed graphs as well. According to this deterministic result, all that matters is the total rate 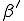 at which an infected node transmits infection to other nodes - not the details of how it distributes that infection.
at which an infected node transmits infection to other nodes - not the details of how it distributes that infection.
As we have just seen, the deterministic approximation is useful for estimating some important characteristics of epidemics - the conditions under which they occur, the rate at which they grow, and the expected number of infections once they have reached equilibrium. However, since it ignores the stochastic nature of an epidemic, it provides no information about other important features of the dynamics, including the size of fluctuations in the number of infected individuals and the possibility that fluctuations will result in extinction of the infection. Consider for a moment the issue of the survival of the virus in a population. The deterministic analysis concludes that there will be an epidemic if and there will not be one if 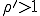 . However, it is intuitively clear that, even if , a statistical fluctuation might wipe out the virus before it spreads to enough individuals to become firmly established. With a little more effort, we can formulate an approximate probahilistic analysis which captures these and certain other important aspects of epidemics which can not be obtained from a deterministic analysis.
. However, it is intuitively clear that, even if , a statistical fluctuation might wipe out the virus before it spreads to enough individuals to become firmly established. With a little more effort, we can formulate an approximate probahilistic analysis which captures these and certain other important aspects of epidemics which can not be obtained from a deterministic analysis.
As in the deterministic analysis of the previous section, we shall assume that the number of infected nodes sufficiently characterises the state of a system, i.e., the details of which nodes are infected are relatively unimportant. Although it is very easy to construct particular graphs for which such details are important (e.g., small graphs with a large variation in the in-degree and out-degree of nodes), we assume that the properties of most members of the class of random graphs with N nodes and edge probability p will not be sensitive to them. Again, we must resort to simulation (in section 2.3) to test the validity of these assumptions. First, we shall describe the probabilistic approximation. Then, we shall use it to calculate various quantities of interest.
Let p(I,t) denote the probability distribution for there to be I infected individuals at time t. Many quantities of interest can be calculated from this distribution. The probability that the infection is extinct at time t is represented by p(0,t), and the expected number of infected individuals and its variance are easily computed by appropriate sums over the p(I,t). The time-evolution of this distribution is given by:
![\frac{dp(I,t)}{dt}=-p(I,t)[R_{I\rightarrow I_+} + R_{I\rightarrow I_-}] + p(I_+,t)R_{I+\rightarrow I} + p(I_-,t)R_{I_-\ \rightarrow I}](../img/cache/04b826221c1f97c3f47c9a9a370c5506.gif) , (3)
, (3)
where 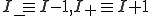 , and
, and 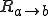 denotes the rate at which transitions occur from state a to state b.
denotes the rate at which transitions occur from state a to state b.
The rates can be calculated as follows. The probability per unit time that a new infedion will occur is simply the number of infected nodes times the rate at which each node tries to infect each of its neighbors times the probability that a given neighbor is susceptible times the number of neighbors. If we assume that the various probabilities are independent (i.e., there is no correlation between the probability that a node is infected and the probability that its neighbors are infected), we obtain:
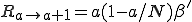 . (4)
. (4)
where is the average total rate at which a node attempts to infect its neighbors. The probability per unit time that an infected node will be cured is simply the number of infected nodes times the rate at which each is cured:
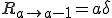 . (5)
. (5)
Substituting these approximations for the rates into Eq. 3, we obtain:
![\frac{dp(I,t)}{dt}=-p(I,t)[I(1-i)\beta'+\delta I]+p(I_+,t)I_+\delta+p(I_,t)[I_-(1-i_-)\beta']](../img/cache/28825bc24c883745b4b932fc920cd51c.gif) , (6)
, (6)
where 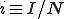 and
and 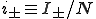 . For a graph with N nodes, this is a set of N + 1 coupled linear differential equations which is relatively simple to solve because the matrix is tridiagonal.
. For a graph with N nodes, this is a set of N + 1 coupled linear differential equations which is relatively simple to solve because the matrix is tridiagonal.
Figure 2 compares the expected number of infected individuals as a function of time as obtained from Eq. 6 with the deterministic result given by Eq. 2. The graph contains 100 nodes with connectivity 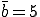 , and the infection and cure rates are
, and the infection and cure rates are 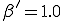 and
and 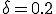 , respectively4. The agreement between the deterministic and stochastic averages is quite good, except for a notable difference between t = 7 and t = 12, when the number of infections starts to saturate. The expected magnitude of the stochastic deviations from run to run, represented by the gray area, grow to a maximum of ±20 at t = 6.3 and then diminish to ±4.51 in equilibrium. The large deviations during the exponential rise can be attributed to the extreme sensitivity of the number of infected individuals at a particular time to random jitter in when the exponential rise occurs. Thus, one would expect a lot of variance in the number of infected individuals from one simulation run to another. However, in equilibrium the size of the infected population is completely insensitive to the moment at which the exponential rise occurred, and the variations from one run to another are much smaller. In equilibrium, the ergodic hypothesis [33] also allows us to interpret these variations as the magnitude of fluctuations about the equilibrium.
, respectively4. The agreement between the deterministic and stochastic averages is quite good, except for a notable difference between t = 7 and t = 12, when the number of infections starts to saturate. The expected magnitude of the stochastic deviations from run to run, represented by the gray area, grow to a maximum of ±20 at t = 6.3 and then diminish to ±4.51 in equilibrium. The large deviations during the exponential rise can be attributed to the extreme sensitivity of the number of infected individuals at a particular time to random jitter in when the exponential rise occurs. Thus, one would expect a lot of variance in the number of infected individuals from one simulation run to another. However, in equilibrium the size of the infected population is completely insensitive to the moment at which the exponential rise occurred, and the variations from one run to another are much smaller. In equilibrium, the ergodic hypothesis [33] also allows us to interpret these variations as the magnitude of fluctuations about the equilibrium.
The same dynamics are presented from a different point of view in Figure 3, which shows snapshots of p(I,t) at successive stages in its evolution. The parameters are the same as in Fig. 2. Initially, at time t = 0, the probability distribution is a delta function at I = 1 (i.e., there is exactly one infected node). As time passes, the probability distribution splits into two components: a delta function at I = 0 (corresponding to extinction of the virus) and a “survival” component which is initially distributed exponentially (t = 1). At first, the mean of the survival component increases exponentially in time, and the standard deviation grows quite large, reaching a maximum of 20 at t = 6.3. Soon, however, the population becomes saturated with infected individuals, and the survival component is nearly gaussian with a mean of 79.75 and a standard deviation of 4.51 at t = 20. This “metastable” phase is extremely long-lived, but the extinction component grows extremely slowly at the expense of the survival component until finally it is all that remains. In general, any population of viruses will eventually die out, but the time scale on which this takes place is so long as to be unobservable unless the graph is quite small. Eventual extinction is inevitable because there is a very tiny probability that all infected individuals will detect and cure their infection at approximately the same time.
The fact that the metastable phase has a finite lifetime
means that we cannot define the probability that the virus becomes extinct without specifying the time period of interest. However, in practice the choice of a “time limit” has little effect on the measured extinction probability provided that it is somewhere within the wide timespan of the metastable regime. For those epidemics which have not died out by a certain time limit, we are interested in the form of the survival component - the distribution p(I,t) for I > 0. Although p(I,t) itself approaches 0 as 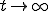 , the conditional probability for there to be I infections given that there is at least one infection approaches a well-defined metastable distribution:
, the conditional probability for there to be I infections given that there is at least one infection approaches a well-defined metastable distribution:
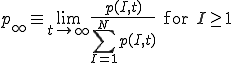 . (7)
. (7)
The survival component is then
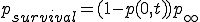 . (8)
. (8)
Given that an epidemic is still active after a given amount of time, we can calculate from 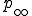 the expected number of infected individuals and the fluctuations about that expectation, and these quantities approach a well-defined asymptote.
the expected number of infected individuals and the fluctuations about that expectation, and these quantities approach a well-defined asymptote.
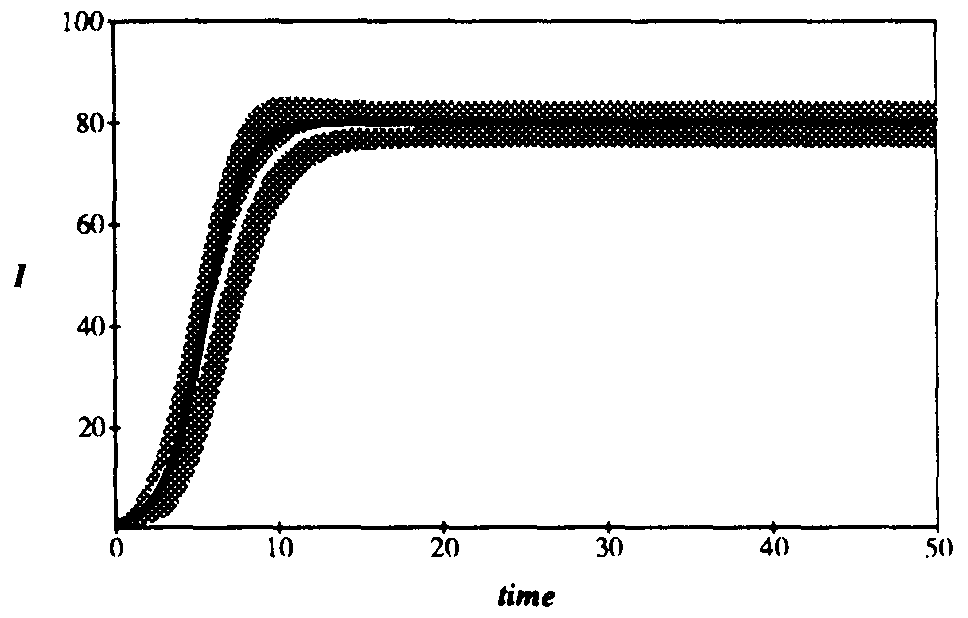
Figure 2: Comparison of the number of infected nodes I as a function of time in the deterministic and stochastic models. The total population is 100 nodes. The average rate at which a node attempts to infect its neighbors is , and the cure rate is . Thus the system is above the classical threshold for an epidemic by a factor of 5. Black curve: deterministic I(t). White curve: stochastic average of I(t). Gray area: One standard deviation about the stochastic average. The final equilibrium values differ by only 0.3%. For t > 15, the gray area can be interpreted as the magnitude of fluctuations about the equilibrium.
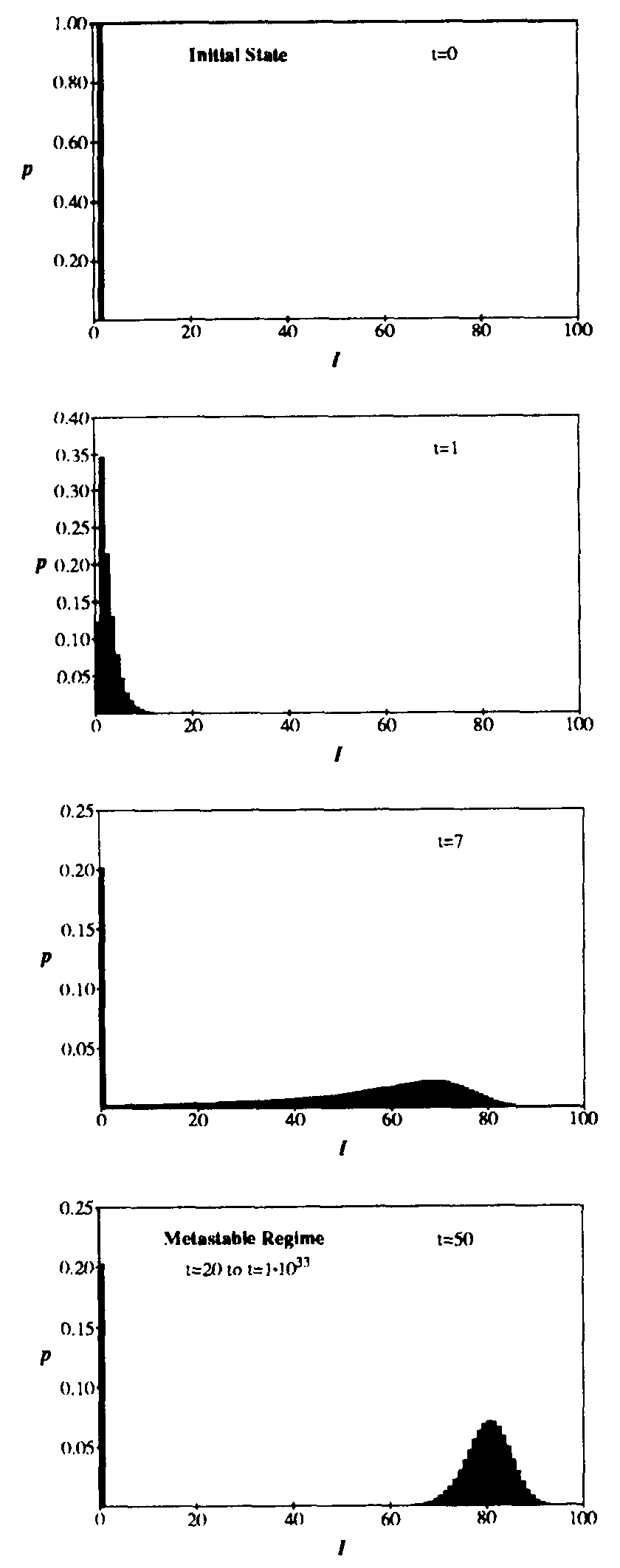
Figure 3: Evolution of the probability distribution for the number of infected individuals in the stochastic approximation. All parameters are the same as in Fig. 2. Starting from a state in which one individual is infected at t = 0, the distribution splits into an “extinction” component (I = 0) and a “survival component” which eventually assumes a gaussian form with the same average and standard deviation as in Fig. 2. The survival component lasts for an extremely long time, but decays with a time constant of 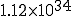 . Note: vertical scales are not all the same.
. Note: vertical scales are not all the same.
By making a few more approximations, we can derive expressions for the probability of extinction as a function
of time, the expected lifetime of the epidemic, and the form of the long-lived metastable distribution .
First, we shall derive an approximate expression for . Setting the derivative on the left-hand side of Eq. 6 equal to zero, we obtain an equation for the equilibrium distribution 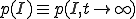 :
:
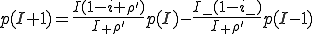 , (9)
, (9)
where p(I)=0 for I < 0 or I > N and 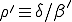 . Substituting I = 0 into Eq. 9, we obtain p(1) = 0. We can then substitute p(1) = 0 into the equation to obtain p(2) = 0. Continuing in the same fashion, we can show trivially that p(I) = 0 for all I > 0. This demonstrates our previous claim that the only equilibrium solution is the extinction component.
. Substituting I = 0 into Eq. 9, we obtain p(1) = 0. We can then substitute p(1) = 0 into the equation to obtain p(2) = 0. Continuing in the same fashion, we can show trivially that p(I) = 0 for all I > 0. This demonstrates our previous claim that the only equilibrium solution is the extinction component.
However, the survival component is nearly a solution, and is only prevented from being one because of the slow leakage of probability to the extinction component. We may obtain the survival component by artificially stopping this leakage, which is achieved by setting p(I = 1) to some arbitrary constant 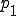 and using Eq. 9 to generate p(I) for I > 1. The resultant distribution - the survival component - should then be normalized such that the sum of the probabilities is unity. Carrying out this procedure with , one can show that the survival component has an extremely short lifetime, which is consistent with the conclusion of the deterministic analysis that no epidemic can occur if the infection rate is less than the cure rate. On the other hand, if
and using Eq. 9 to generate p(I) for I > 1. The resultant distribution - the survival component - should then be normalized such that the sum of the probabilities is unity. Carrying out this procedure with , one can show that the survival component has an extremely short lifetime, which is consistent with the conclusion of the deterministic analysis that no epidemic can occur if the infection rate is less than the cure rate. On the other hand, if 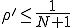 , one can show that practically all of the nodes will be infected, and the lifetime of the survival component will be extremely long.
, one can show that practically all of the nodes will be infected, and the lifetime of the survival component will be extremely long.
In the more interesting intermediate case in which 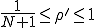 , p(I) attains a maximum for some
, p(I) attains a maximum for some 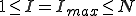 . The value of
. The value of 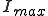 is determined by the condition
is determined by the condition 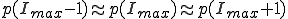 . Substitution of this condition into Eq. 9 yields
. Substitution of this condition into Eq. 9 yields
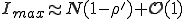 . (10)
. (10)
Motivated by the form of the survival component in the last two frames of Fig. 3, we match a gaussian to 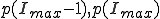 , and
, and 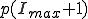 and normalize the sum of the extinction and survival components to unity, with the result:
and normalize the sum of the extinction and survival components to unity, with the result:
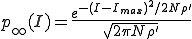 . (11)
. (11)
Thus we find that the metastable distribution is a gaussian with mean 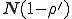 and standard deviation
and standard deviation 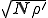 . The mean is identical to that obtained from the deterministic approximation, and the standard deviation is virtually equal to that obtained from the numerical solution of Eq. 6 depicted in Fig. 3.
. The mean is identical to that obtained from the deterministic approximation, and the standard deviation is virtually equal to that obtained from the numerical solution of Eq. 6 depicted in Fig. 3.
Having obtained the metastable distribution, we can now use it to estimate the lifetime of the metastable survival component. First, we must obtain an expression for p(0,t), the extinction probability as a function of time. Returning to the dynamical equation for the evolution of the probability distribution (Eq. 6), substituting I = 0, and using Eqs. 8 (which is valid once the distribution has assumed its metastable form) and 11, we obtain:
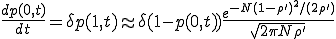 . (12)
. (12)
Solving Eq. 12 for p(0,t) , we find that, after the initial transient, the survival component decays exponentially with a characteristic lifetime given by
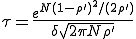 . (13)
. (13)
Numerical solution of Eq. 9 reveals that Eq. 13 yields a rather severe overestimate of the lifetime of the metastable phase. This can be attributed to the fact that, while the gaussian approximation of Eq. 11 to the survival component is very good in the vicinity of , it is exceedingly poor in the far reaches of the tail, at I = 1. However, the numerical solution confirms that the functional form of Eq. 13 is correct, i.e., the lifetime of a graph increases approximately exponentially with the number of nodes. For example, the lifetime of for a 100-node graph is reduced to only 888 for a 10-node graph, all other parameters being equal. The constant multiplying N in the exponent is about half that predicted by Eq. 13 when 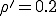 .
.
Finally, we can obtain a good approximation to the probability that the infection will become extinct before it reaches the metastable phase by letting the number of nodes 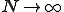 do (rendering the lifetime infinite). Then Eq. 6 simplifies to:
do (rendering the lifetime infinite). Then Eq. 6 simplifies to:
![\frac{dp(I,t)}{dt}=-p(I,t)[I(\beta'+\delta)]p(I_+,t)[I_+\delta]+p(I_-,t)[I_-\beta']](../img/cache/a4f75fd9b1d56e28d5e0046d19560dfa.gif) , (14)
, (14)
which is the well-known linear birth and death process [34]. A solution can be obtained by the method of generating functions, with the result:
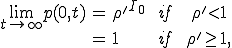 , (15)
, (15)
where 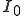 is the number of infected nodes which are originally present in the population.
is the number of infected nodes which are originally present in the population.
To summarize, the probabilistic analysis corroborates the deterministic result that an epidemic can not occur if 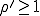 . Contrary to the findings of the deterministic
analysis, even if
. Contrary to the findings of the deterministic
analysis, even if 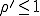 , the probability that an epidemic will not occur is greater than zero, being equal to
, the probability that an epidemic will not occur is greater than zero, being equal to 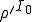 , where is the number of infected nodes initially present in the population. This is not really a discrepancy. The deterministic analysis is founded on the assumption of an infinite number of nodes and an original fraction of infected nodes . Thus is infinite, and the probabilistic formula (Eq. 15) yields an extinction probability of zero. The probabilistic and deterministic analyses agree that the average number of infected nodes in equilibrium is , and the probabilistic analysis reveals that the root-mean-square fluctuations are
, where is the number of infected nodes initially present in the population. This is not really a discrepancy. The deterministic analysis is founded on the assumption of an infinite number of nodes and an original fraction of infected nodes . Thus is infinite, and the probabilistic formula (Eq. 15) yields an extinction probability of zero. The probabilistic and deterministic analyses agree that the average number of infected nodes in equilibrium is , and the probabilistic analysis reveals that the root-mean-square fluctuations are 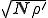 . Thus the relative size of the fluctuations decreases as
. Thus the relative size of the fluctuations decreases as 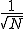 , which lends some justification to the assumption that they are zero in the deterministic analysis.
, which lends some justification to the assumption that they are zero in the deterministic analysis.
Both the deterministic and stochastic analyses of the model required a number of assumptions which can only be tested by simulation. We have simulated the model using a straightforward event-driven implementation. A graph is generated randomly according to the prescription given at the beginning of this section, and a single initially-infected node is selected randomly. Then, the simulation proceeds one event (i.e., an attempted infection or cure of a node) at a time, using time steps generated randomly according to an exponential distribution5. The mean of this distribution is determined by the probabilities per unit time of infection and cure, the number of infected nodes, and the number of edges emanating from the collection of infected nodes.
Figure 4 compares a typical simulation run on a 100-node graph to the corresponding deterministic solution, using the parameters of Figs. 2 and 3. The simulation run follows the deterministic solution reasonably well, except that the equilibrium appears to be lower.
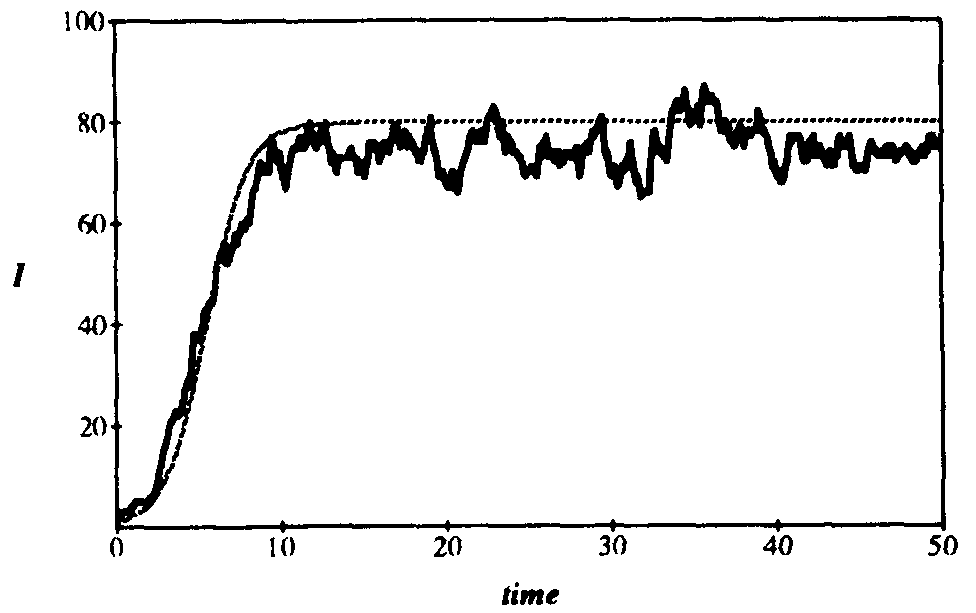
Figure 4: Comparison between average number of infected individuals vs. time as given by deterministic theory and a typical simulation run on a randomly-generated graph with 100 nodes. The average number of edges emanatingfrom each node is 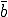 = 5, and all other parameters are as givenin Fig. 2. The magnitude of the fluctuations agrees reasonably well with that predicted by the stochastic theory (compare with Fig. 2), but the average number of infected individuals is slightly lower. This discrepancy can be attributed to the low connectivity of the graph ( = 5).
= 5, and all other parameters are as givenin Fig. 2. The magnitude of the fluctuations agrees reasonably well with that predicted by the stochastic theory (compare with Fig. 2), but the average number of infected individuals is slightly lower. This discrepancy can be attributed to the low connectivity of the graph ( = 5).
To investigate this discrepancy, we performed 2500 simulation runs using the same parameters but different seeds for the random number generator. In 25.9±0.9% of the runs, the population became extinct by t = 1200 (a time limit that we chose to be comfortably within the metastable regime). This is noticeably larger than the extinction probability of 0.20 predicted by Eq. 15. For each of those runs which survived, we measured the average number of infected individuals between t = 200 and t = 1200 and the fluctuations about this equilibrium. The average equilibrium for these runs was 75.01±0.04, which as we suspected from Fig. 4 is significantly lower than the deterministic prediction of 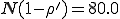 and the stochastic prediction of 79.75. The average magnitude of the fluctuations within a run was 4.857±0.005, somewhat larger than the stochastic prediction of 4.508. The variation in the equilibrium obtained across different simulation runs was only ±1.65, indicating that the entire class of random graphs is in fact well-characterized by these measurements - one of the main assumptions made in the deterministic and stochastic analyses.
and the stochastic prediction of 79.75. The average magnitude of the fluctuations within a run was 4.857±0.005, somewhat larger than the stochastic prediction of 4.508. The variation in the equilibrium obtained across different simulation runs was only ±1.65, indicating that the entire class of random graphs is in fact well-characterized by these measurements - one of the main assumptions made in the deterministic and stochastic analyses.
Why is the extinction probability a bit higher and the average number of infected individuals a bit lower than predicted? The fault must lie in one or more of the approximations that were used to derive the deterministic and stochastic theories. The most likely suspect is the neglect of the particular details of how nodes are connected to one another. This assumption came into play in at least two guises. First, it allowed us to assume that the dynamics could be expressed solely in terms of how many nodes were infected, without having to delve into the details of which were infected (a problem that would be completely intractable). Second, we neglected variation in the number of nodes that a given node could ii:fect, assuming that every node tried to infect exactly other nodes. Intuitively, we expect both of these assumptions to become increasingly valid as the connectivity is increased.
Imagine for a moment the extreme limit of tenuousness (below what is referred to by random graph theorists as the percolation threshold [35]), in which most nodes are isolated and a few are joined in small clusters. It is readily apparent that infection cannot spread beyond the small cluster in which the initially infected individual is located. Thus the equilibrium level should be depressed substantially below the homogeneous limit. If the infection is confined to very small clusters, it becomes much more likely that all infections in the cluster will be detected and cured at approximately the same time. In such a case, the lifetime of the metastable phase could become less than our chosen time limit, in which case the measured extinction probability would increase. Thus, infections should die out more easily in tenuous graphs.
This conjecture is borne out by Figure 5, in which we have varied the connectivity of the graph , keeping the average total rate at which a node-attempts to infect its neighbors fixed at = 1.0. For < 1.0, it is nearly certain that the infection will die out quickly. Tke extinction probability drops precipitously for 1 < < 2 and slowly approaches the homogeneous limit of 0.20 as is increased to 10 and beyond. When the simulations are repeated on graphs of 1000 rather than 100 nodes, the behavior is quite similar, except that the transition in the range 1 < < 2 becomes slightly sharper. In Fig. 5b, we see that the average number of infections in equilibrium is severely depressed below the homogeneous limit in tenuous graphs. Again, there is a characteristic curve which is fairly insensitive to the size of the graph except in the transition region, which becomes sharper for larger graphs. We have observed similar qualitative behavior in simulations in which 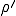 = 0.5, in which case the tracsition region is shifted upwards to approximately 2 < < 4.
= 0.5, in which case the tracsition region is shifted upwards to approximately 2 < < 4.
In the random graph model that we have examined so far, a node is able to infect only a few other nodes in the graph (at least for the typical situation in which 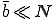 ). Although it is probably true that most users share most of their programs with a small number of other individuals, there is generally a much larger group of other individuals with whom they exchange programs every once in a while. To what extent will these extra pathways enable viruses to spread?
). Although it is probably true that most users share most of their programs with a small number of other individuals, there is generally a much larger group of other individuals with whom they exchange programs every once in a while. To what extent will these extra pathways enable viruses to spread?
We study the effect of infrequent sharing with a large number of other individuals by modifying the random graph model slightly, giving a node a small but finite chance of infecting any node which is not explicitly connected to it. In addition to the previously-defined infection rate 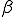 , which we shall now refer to as the "strong" infection rate, we define the "weak" infection rate
, which we shall now refer to as the "strong" infection rate, we define the "weak" infection rate 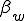 . As before, the average total-infection rate through the "strong" links is given by
. As before, the average total-infection rate through the "strong" links is given by 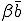 . The average total infection rate through the "weak" links is
. The average total infection rate through the "weak" links is 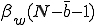 . Thus the total infection rate through all links is:
. Thus the total infection rate through all links is:
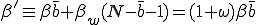 , (16)
, (16)
where
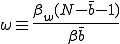 (17)
(17)
is the ratio between the total weak and strong infection rates.
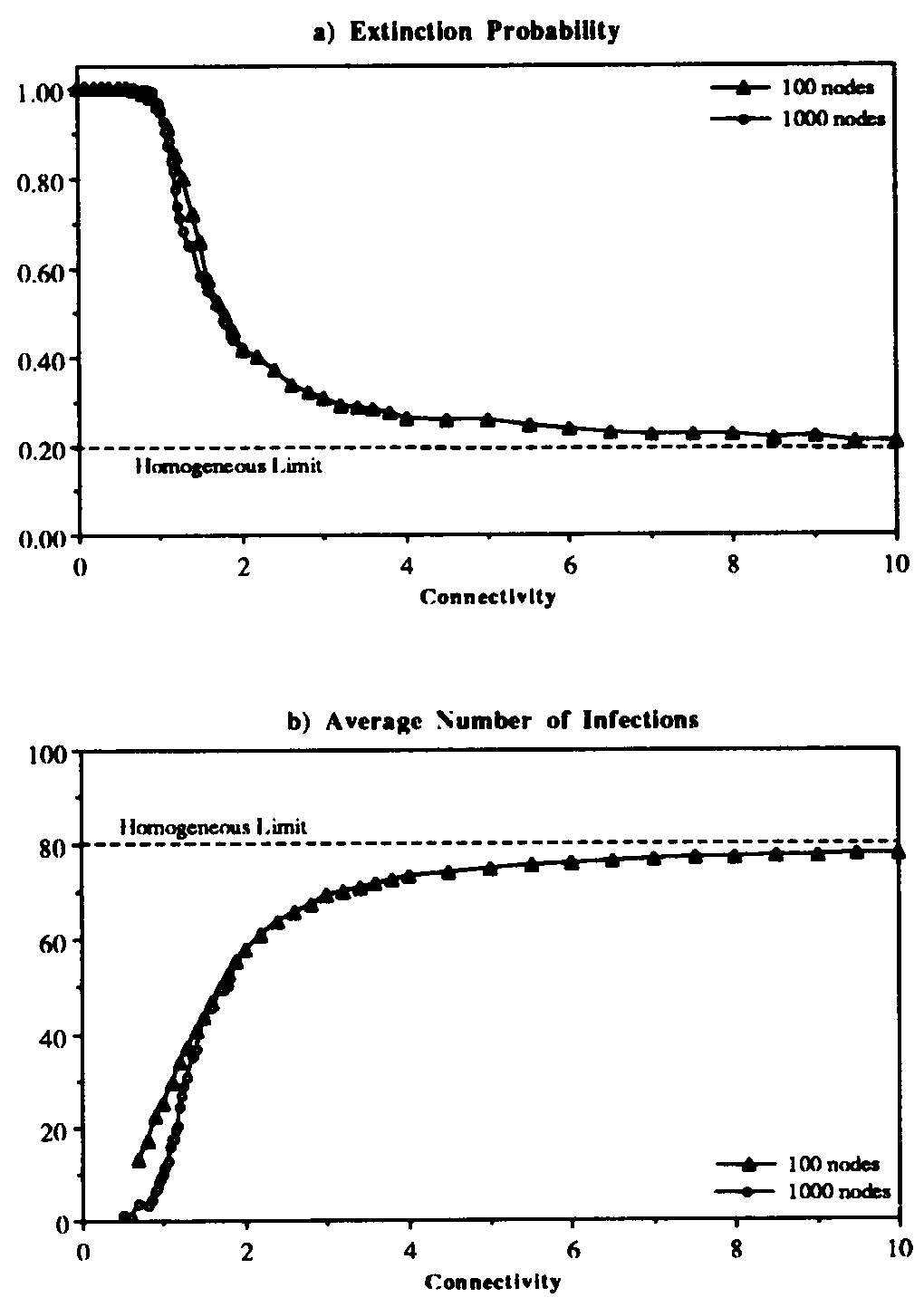
Figure 5: Extinction probability (a) and average number of infections (b) vs. connectivity for random graph with the usual infection and cure rates: and . Each point represents an average over 2500 simulation runs. For < 1, it was extremely rare for an epidemic to survive beyond the time limit of 1200, despite the fact that the infection rate is 5 times the classical epidemic threshold. For higher connectivities, the extinction probability and the average number of infected individuals approach the values predicted by the classical homogeneous interaction theory. The dependence of these quantities upon the connectivity changes very little when the number of nodes in the graph is increased from 100 to 1000; the transition region becomes slightly sharper. (Note: the measured equilibria for 1000-node graphs have been divided by 10 to scale them properly to the 100-node results.)
Figure 6 displays the effect upon the extinction probability and the average number of infections in equilibrium of various ratios  . The results for
. The results for  are simply those of Fig. 5, i.e., there are no weak links. When is increased to 0.2, the qualitative behavior of both the extinction probability and the average number of infections is the same, but the transition region is shifted towards more tenuous connectivities. For example, a graph with = 0.9, which is practically impervious to infection when there are no weak lipks, becomes behaviorally equivalent to a graph with = 1.5: the extinction probability drops to nearly 1/2, and nearly half of the population is infected in equilibrium. When is increased further to 0.5, the transition region disappears.
Thus for
are simply those of Fig. 5, i.e., there are no weak links. When is increased to 0.2, the qualitative behavior of both the extinction probability and the average number of infections is the same, but the transition region is shifted towards more tenuous connectivities. For example, a graph with = 0.9, which is practically impervious to infection when there are no weak lipks, becomes behaviorally equivalent to a graph with = 1.5: the extinction probability drops to nearly 1/2, and nearly half of the population is infected in equilibrium. When is increased further to 0.5, the transition region disappears.
Thus for 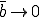 there is a finite probability (0.40) for an epidemic to occur, and the average number of infected individuals in equilibrium is 40. These limiting values are easily understood by completely disregarding the strong links, in which case we have from Eq. 16 that the effective value of is 1/3 of its nominal value, or
there is a finite probability (0.40) for an epidemic to occur, and the average number of infected individuals in equilibrium is 40. These limiting values are easily understood by completely disregarding the strong links, in which case we have from Eq. 16 that the effective value of is 1/3 of its nominal value, or 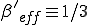 . Since
. Since 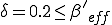 , the conditions for an epidemic to occur are satisfied by the weak links alone, with an effective value of
, the conditions for an epidemic to occur are satisfied by the weak links alone, with an effective value of 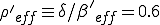 , in agreement with the simulation.
, in agreement with the simulation.
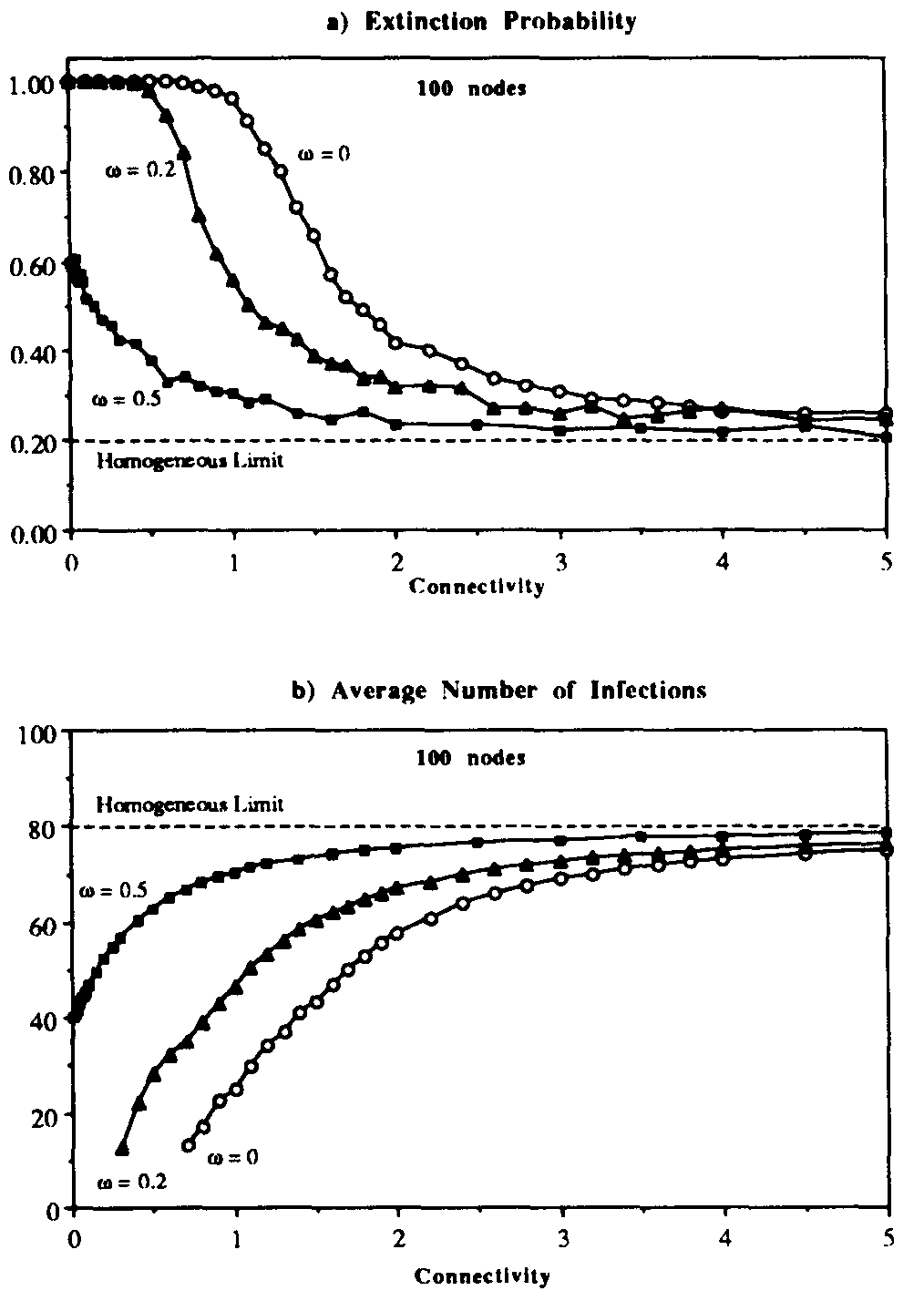
Figure 6: Extinction probability (a) and average number of infections (b) vs. connectivity for random graph with weak links. Three different values of the ratio between the sum of the weaklink infection rates and the sum of the strong-link infection rates are presented. As usual, the infection and cure rates arc = 1.0 and = 0.2. Each point represents an average over 2500 simulation runs on 100-nodegraphs. As the weak links become stronger, the extinction probability and the average number of infections approach their homogeneous limits over a wider range of connectivities.
The simulation results of Figs. 5 and 6 suggest that the homogeneous limit is reasonably good when the total infection rate is spread among a sufficient number of nodes. However, when too much of the total infection rate is concentrated into too few nodes, epidemics have a much harder time establishing themselves than would be predicted by the homogeneous approximation. For example, according to Fig. 6a, when the connectivity = 1 and there are no weak links, the epidemic threshold is approximately 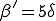 , which is five times the classical value. Other simulations that we have not presented here indicate that, for = 2, the epidemic threshold is about twice the classical value. These results are unaffected by the number of nodes N in the graph, provided that N exceeds about 100. Weak links diminish the increase in the threshold, but do not eliminate it. For example, when the total infection rate through the weak links is 0.2 times the total infection rate through the strong links ( = 0.2), the threshold can be five times the classical value, but only if the graph is twice as tenuous ( = 0.5) as it needs to be when there are no weak links.
, which is five times the classical value. Other simulations that we have not presented here indicate that, for = 2, the epidemic threshold is about twice the classical value. These results are unaffected by the number of nodes N in the graph, provided that N exceeds about 100. Weak links diminish the increase in the threshold, but do not eliminate it. For example, when the total infection rate through the weak links is 0.2 times the total infection rate through the strong links ( = 0.2), the threshold can be five times the classical value, but only if the graph is twice as tenuous ( = 0.5) as it needs to be when there are no weak links.
Motivated by a desire to study the effect of infrequent program sharing with many other individuals, we augmented the random graph model by introducing weak links in section 2.4. However, the classification of links into just two types - strong and weak - is a bit crude. It is probably more realistic to assume that an individual exchanges programs at a very high rate with very few other individuals, at a lower rate with a larger class of other individuals, at an even lower rate with an even larger class of others, etc. This leads naturally to the hierarchical model illustrated in Figure 7.
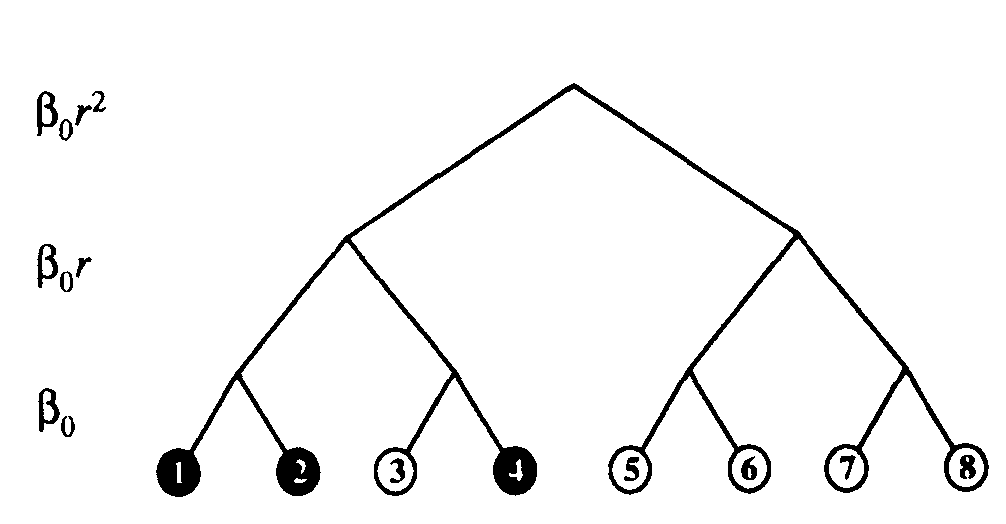
Figure 7: Binary hierarchical graph with 3 levels. Infection rates between nodes separated by a given number of levels are listed to the left of the tree. Infected and uninfected nodes are represented by black and unfilled circles, respectively. Initially, only node 2 was infected, but it quickly infected node 1. For a while, nodes 1 and 2 attempted to infect one another (to no effect, since they were already infected). Eventually, node 1attempted successfully to infect node 3. Node 3 quickly infected node 4, but was cured soon afterward. Node 3 will probably be infected soon by node 4, but nodes 5, 6, 7, and 8 will probably not be infected for a relatively long time.
The hierarchical model has an unexpected side benefit. Note that all nodes with a given subtree communicate with one another more frequently than with any node in any other subtree. Thus, the hierarchy of rates automatically enforces a hierarchy of cliques6. In the computer community, cliques of users certainly exist, although not in such an extreme form as in this model. For example, users within a department may share software frequently among themselves, less frequently with users in other departments of the same company or university, and still less frequently with users in other cities or countries. The random graph model and others derived from it are incapable of accounting for such correlations because of the way in which they are constructed - the connections are chosen randomly and independently.
For simplicity, we assume that the frequency of contact between two nodes decreases geometrically with the distance 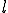 between them (the number of levels one must go up in the tree to reach a common ancestor), while the size of the class of neighbors at a given distance grows geometrically with . More explicitly, assume that the hierarchy consists of a binary tree with L levels. Then there are
between them (the number of levels one must go up in the tree to reach a common ancestor), while the size of the class of neighbors at a given distance grows geometrically with . More explicitly, assume that the hierarchy consists of a binary tree with L levels. Then there are 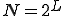 nodes. Suppose that the infection rate between nodes separated by a distance is given by
nodes. Suppose that the infection rate between nodes separated by a distance is given by 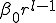 .The number of nodes at distance is simply
.The number of nodes at distance is simply 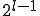 . Therefore, the total infection rate from one node is
. Therefore, the total infection rate from one node is
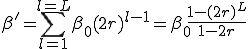 (18)
(18)
As before, we assume that the cure rate for each node is .
By keeping and fixed and varying the localization parameter r, we can explore a wide range of situations. When r = 0, the network effectively consists of isolated pairs of nodes with infection rate . By setting r = 1, we obtain the homogeneous limit, in which the infection rate between all pairs of nodes is equal to 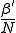 . Thus, the parameter r ought to be similar in its effect to the connectivity .
. Thus, the parameter r ought to be similar in its effect to the connectivity .
We have investigated the dependence of the extinction probability upon the localization parameter by means of simulation. The results are summarized in Fig. 8. As expected, the extinction probability exhibits threshold behavior qualitatively similar to that of Fig. 5a. For strongly localized graphs, extinction of the infected population is virtually guaranteed, even though the infection rates are well above the classical threshold (by a factor of five for 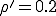 and a factor of two for
and a factor of two for 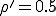 ). As
). As 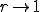 , the extinction probability aproaches the homogeneous limit, as expected. The upward shift in position and the increase in width of the transition region as is increased are both consistent with what we have observed for larger values of as the connectivity is varied in the random graph model.
, the extinction probability aproaches the homogeneous limit, as expected. The upward shift in position and the increase in width of the transition region as is increased are both consistent with what we have observed for larger values of as the connectivity is varied in the random graph model.
Note that we have not presented the corresponding curves for the average number of infected individuals in equilibrium. The reason is quite interesting. When r is in the transition region, the individual simulation runs often fail to attain a well-defined equilibrium. A typical example, shown in Fig. 9, displays a character much more mercurial than that of a typical simulation in the random graph model (Fig. 4). In Fig. 4, a welldefined equilibrium level is reached by t = 10. However, in Fig. 9, the number of infected individuals I varies over a large range in a very irregular fashion even after many hundreds of time units. The range within which I varies lies far below the homogeneous limit of 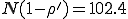 , as is expected since r is in the transition region. Inspection of a number of individual simulation runs strongly suggests that the metastable phase, if it exists, is much shorter in duration than its random graph counterpart. This is reasonable because rapid extinction is consistent with the small number of infected individuals and the large magnitude of the fluctuations in that quantity.
, as is expected since r is in the transition region. Inspection of a number of individual simulation runs strongly suggests that the metastable phase, if it exists, is much shorter in duration than its random graph counterpart. This is reasonable because rapid extinction is consistent with the small number of infected individuals and the large magnitude of the fluctuations in that quantity.
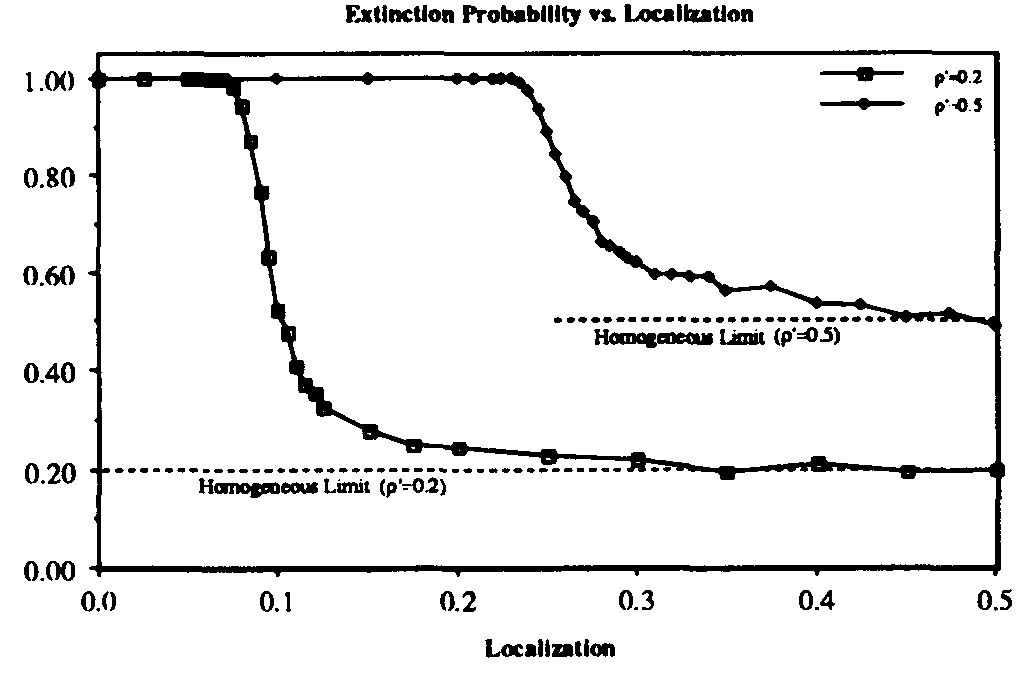
Figure 8: Extinction probability vs. localization parameter r for SIS model on hierarchical graph of 128 nodes. For sufficiently localized interactions (small r), rapid extinction of the virus is virtually assured, even when the infection rate is well above the classical epidemic threshold (by a factor of 5 for = 0.2 and a factor of 2 for = 0.5). The relatively small width of the transition region is reminiscent of Fig. 5a.
We have also performed simulations on larger hierarchical graphs with as many as 8192 nodes. Interestingly, in the transition region the average number of infections does not increase with the size of the graph, and the magnitude and irregular character of the fluctuations is unaltered. This contrasts strongly with the behavior of random graphs, for which the number of infected individuals scales linearly with N and the relative size of the fluctuations decreases as 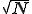 . In some simulation runs, one can identify a series of plateaus in I(t), separated by relatively rapid growth spurts. The growth spurts occur when a node "gets lucky" and infects a distant node, which then spreads the infection throughout a previously untouched region of the graph. When r is sufficiently far above the transition region, I(t) continues to grow until it saturates eventually at the homogeneous equilibrium. The rate at which it does so is much slower than for a random graph and is extremely
sensitive to r (see Fig. 12). The functional form of the growth rate and its dependence upon r is not yet known.
. In some simulation runs, one can identify a series of plateaus in I(t), separated by relatively rapid growth spurts. The growth spurts occur when a node "gets lucky" and infects a distant node, which then spreads the infection throughout a previously untouched region of the graph. When r is sufficiently far above the transition region, I(t) continues to grow until it saturates eventually at the homogeneous equilibrium. The rate at which it does so is much slower than for a random graph and is extremely
sensitive to r (see Fig. 12). The functional form of the growth rate and its dependence upon r is not yet known.
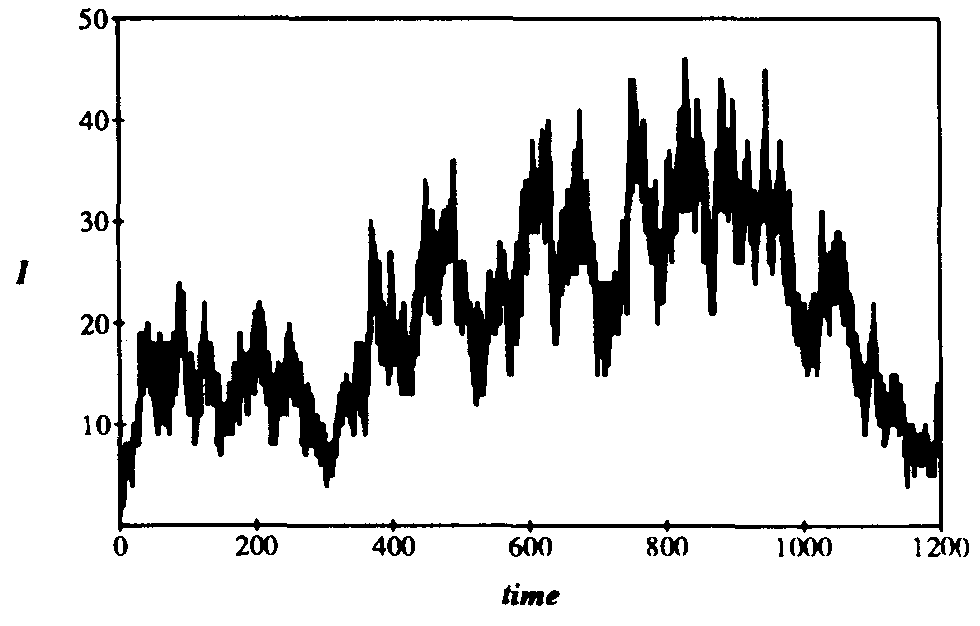
Figure 9: Number of infections I vs. time for typical simulation run on 128-node hierarchical graph with localization parameter r = 0.1. The infection and cure rates have their usual values of = 0.1 and = 0.2. The aimless, irregular drift in the number of infected individuals at a value much lower than the homogeneous equilibrium (I=102.4) is characteristic of simulation runs with localization parameter lying within the transition region (see = 0.2 curve in Fig. 8).
The hierarchical model of the previous section allowed us to explore the consequences of localized program sharing. In this section, we shall learn more about the effects of locality by studying epidemics on a completely different topological structure - a d-dimensional Cartesian lattice. Each point in the lattice represents a node which can infect or be infected by all nodes within some local neighborhood. As in the hierarchical model, locality immediately implies the existence of cliques, but their form is somewhat different in the spatial model. For example, consider the neighborhood of infectible nodes surrounding node A. If we move one step to the rigl-t to node B, we find that B's neighborhood has many nodes in common with A's.
Although it may be less realistic in some respects than the hierarchical model, the spatial model offers the advantage of being more amenable to deterministic analysis. Given our experience that random graphs with sufficient connectivity are reasonably well-described by a deterministic approximation, we expect this to hold for the spatial model as well, provided that the neighborhood is sufficiently large. First, we shall use a deterministic approximation to derive an equation for the spatio-temporal dynamics of an epidemic. Then, we shall confirm these results with simulations.
To begin the analysis, imagine imbedding a graph in a d-dimensional space by associating each node j arbitrarily with a position 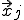 . We can derive a deterministic approximation for
. We can derive a deterministic approximation for 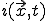 ,the fraction of infected individuals at position
,the fraction of infected individuals at position 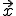 at time t, as follows. Let
at time t, as follows. Let 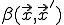 represent the rate at which the node at
represent the rate at which the node at 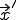 attempts to infect the node at and
attempts to infect the node at and 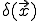 ) represent the rate at which a node at is cured (if it was infected). Then, following the same considerations as were used to derive the deterministic approximation for random graphs, we obtain:
) represent the rate at which a node at is cured (if it was infected). Then, following the same considerations as were used to derive the deterministic approximation for random graphs, we obtain:
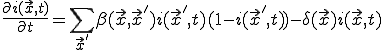 , (19)
, (19)
Note that Eq. 19 is a slightly generalized form of Eq. 1, with node indices replaced by positions.
Now assume that a node can only interact with nodes lying within some small local neighborhood, and that the infection rates between two nodes depend only upon the distance between them. Furthermore, assume that the cure rate is independent of . Then, if and 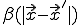 vary sufficiently slowly from one node to another, we can treat them as continuous functions, in which case it can be shown that Eq. 19 becomes approximately:
vary sufficiently slowly from one node to another, we can treat them as continuous functions, in which case it can be shown that Eq. 19 becomes approximately:
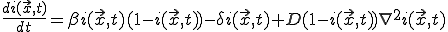 , (20)
, (20)
where
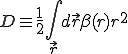 . (21)
. (21)
The first two terms in Eq. 20 are the familiar growth and decay terms that appear in Eq. 1. By themselves, they describe growth or decay of the level of infection at each point in space independently of the dynamics at any other point in space. The last term is something new. It is a second-order spatial derivative which accounts for diffusion of infection between different points in space. The diffusion coefficient D can be derived for various assumptions about the influence of nodes upon their neighbors. For example, if 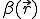 is uniform within a hypercubic volume
is uniform within a hypercubic volume 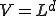 with integral
with integral 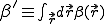 ,
, 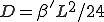 . If is gaussian-distributed with standard deviation
. If is gaussian-distributed with standard deviation 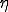 ,
, 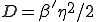 .
.
Due to the assumed radial symmetry of 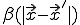 , will remain radially symmetric if the initial condition
, will remain radially symmetric if the initial condition 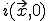 is. Such a choice greatly simplifies both the calculation and the presentation of the results. Figure 10 depicts the typical course of an epidemic in two dimensions as predicted by Eq. 20, where
is. Such a choice greatly simplifies both the calculation and the presentation of the results. Figure 10 depicts the typical course of an epidemic in two dimensions as predicted by Eq. 20, where 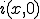 is a narrow gaussian with volume
is a narrow gaussian with volume 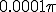 .The population inhabits a circle of radius 1, so this initial distribution constitutes 1/10000 of the population.
.The population inhabits a circle of radius 1, so this initial distribution constitutes 1/10000 of the population.
In its first phase of growth, the pulse grows in height (t = 4). When the pulse saturates at the equilibrium
value of 0.8, it remains pinned at that value but keeps spreading outward radially. The leading front of the expanding circle develops a sharp edge, with a radius that increases at a constant velocity (note the positions at t = 20, 50,and 80). Thus the number of infected individuals, proportional to the area of the circle, increases quadratically with time. In d dimensions, the infection expands outward at constant velocity as a sharp-edged sphere, so the number of infected individuals grows as 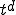 . This is of course much slower than the exponential growth of the random graph model. Finally, when the infection reaches the entire population, the total fraction of infected individuals reaches the same limit as in the random graph model, and its distribution is spatially uniform.
. This is of course much slower than the exponential growth of the random graph model. Finally, when the infection reaches the entire population, the total fraction of infected individuals reaches the same limit as in the random graph model, and its distribution is spatially uniform.
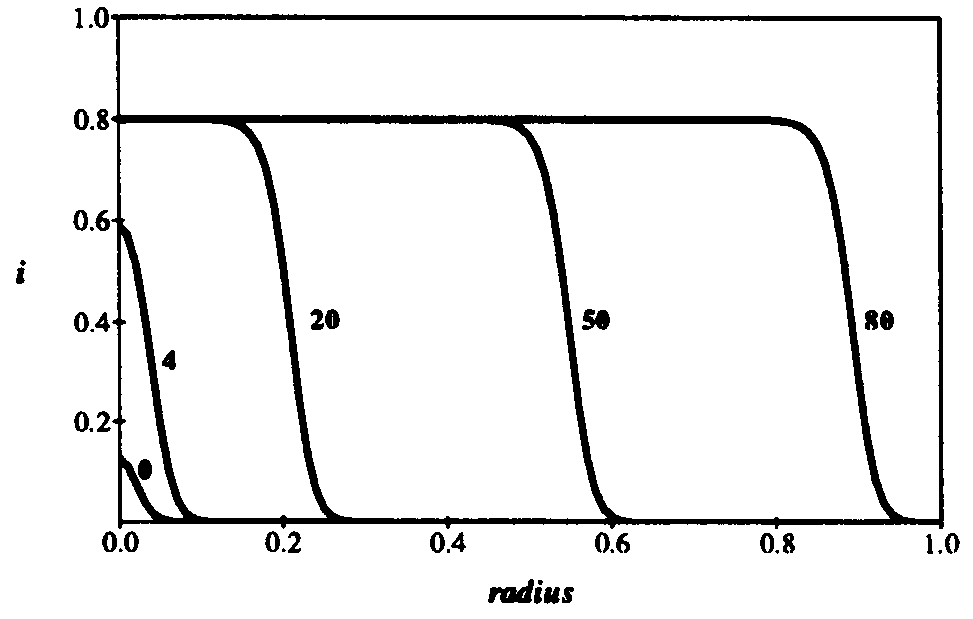
Figure 10: Density of infected individuals i as a function of radius r from the initially source of infection at times t = 0,4, 20, 50, and 80. As usual, the infection and cure rates are = 1.0 and = 0.2. The diffusion coefficient 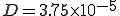 . Initially, 0.0001 of the population is infected, represented by a narrow gaussian with a standard deviation of 0.02 near r = 0. At first, the height of the gaussian grows, until it saturates at the homogeneous limit of 0.8. Then, the infection enters a diffusive phase, growing outward at constant velocity in a circle with a fairly sharp boundary of fixed shape. Eventually, the spatial distribution of infection becomes uniform, with 80% of the individuals being infected.
. Initially, 0.0001 of the population is infected, represented by a narrow gaussian with a standard deviation of 0.02 near r = 0. At first, the height of the gaussian grows, until it saturates at the homogeneous limit of 0.8. Then, the infection enters a diffusive phase, growing outward at constant velocity in a circle with a fairly sharp boundary of fixed shape. Eventually, the spatial distribution of infection becomes uniform, with 80% of the individuals being infected.
We have simulated epidemics on a two-dimensional square array wrapped around in both dimensions to form a torus (so as to avoid edge effects). The neighborhood about each node is an 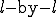 block centered on (but not including) itself. The infection rate is equal to
block centered on (but not including) itself. The infection rate is equal to 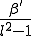 for each node within that neighborhood and zero for all nodes outside of it. The dynamical details of the simulation are identical to those which we have presented for the random graph; the only difference lies in the choice of infection rates between pairs of nodes. Thus, if we were to expand the neighborhood to include all of the nodes, our resultant system would be equivalent to a fully-connected graph and its dynamics would be described by the homogeneous limit.
for each node within that neighborhood and zero for all nodes outside of it. The dynamical details of the simulation are identical to those which we have presented for the random graph; the only difference lies in the choice of infection rates between pairs of nodes. Thus, if we were to expand the neighborhood to include all of the nodes, our resultant system would be equivalent to a fully-connected graph and its dynamics would be described by the homogeneous limit.
Figure 11 illustrates the state of a typical simulation run auring the spreading phase of the epidemic. Initially, the central node in the 100 by 100 array was infected. The size of the neighborhood was chosen to be =3, i.e., it was composed of the 8 nearest neighbors. It is interesting to note that, despite the fact that 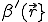 is square in shape, the pattern of infection is roughly circular. In fact, the greater generality of radial spreading is expected from details of the derivation of Eq. 20 which we have omitted here.
is square in shape, the pattern of infection is roughly circular. In fact, the greater generality of radial spreading is expected from details of the derivation of Eq. 20 which we have omitted here.
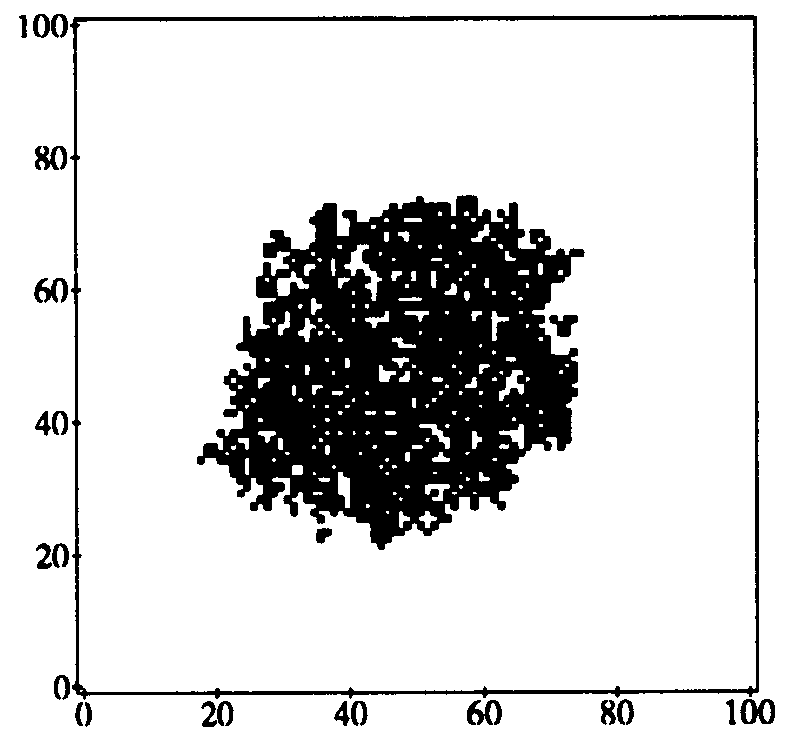
Figure 11: State of an epidemic in diffusion phase (t = 50) as obtained from a simulation on a 100-by-100 array. The infection and cure rates are 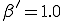 and
and  , as usual. Each node can infect the 8 neighbors lying within a 3-by-3 square centered on itself. Black and white squares represent infected and uninfected individuals, respectively. As in the theoretical curves of Fig. 10, the boundary of the expanding circle of infected nodes is roughly circular and fairly sharp (despite the fact that the infection neighborhood is square).
, as usual. Each node can infect the 8 neighbors lying within a 3-by-3 square centered on itself. Black and white squares represent infected and uninfected individuals, respectively. As in the theoretical curves of Fig. 10, the boundary of the expanding circle of infected nodes is roughly circular and fairly sharp (despite the fact that the infection neighborhood is square).
Simulations verify the quadratic growth with time of the total number of infected individuals. Figure 12 compares the relative rates at which the equilibrium is attained in the two-dimensional lattice with that of the random and hierarchical graphs. In order to provide a fair comparison, the infection and cure rates were their usual values of = 1.0 and = 0.2, and the number of nodes in the simulation of each of the three models was chosen to be as close as possible to 10000. Furthermore, the connectivity of the random graph and the localization parameter of the hierarchical graph were chosen to be well above the transition region, so that the equilibrium would be as close as possible to the homogeneous limit.
It must be emphasized that the curves in Fig. 12 are not to be taken as an absolute comparison of the growth rates for these three models. The exact rates depend upon the diffusion coefficient in the spatial model and are extremely sensitive to the localization parameter in the hierarchical model. (For example, when r is increased by only 10%, the growth rate is increased by about 35%.) It is the functional form of the growth that is of the greatest importance. As expected, the random graph attains the equilibrium at an exponential rate, while the two-dimensional spatial model reaches it at a much slower, quadratic rate. The functional form of the growth rate for the hierarchical graph is not yet known, but in this and other simulations it appears to be quite slow.
Thus both of the local models that we have studied exhibit very slow growth compared to the random graph model (e.g., polynomial rather than exponential in time). We expect that this will prove to be the case for other more complicated and realistic models that take locality into account. In local models, the majority of the infected nodes find themselves in a region that has already reached local equilibrium. It is only the relatively small number of nodes on the expanding front of the infected region that can spread the infection to the uninfected nodes lying outside the region. Since they can only infect nodes lying close by, the vast majority of uninfected nodes lying outside the region are unavailable. The spread of infection on a random graph is much more efficient because the boundary of the infected region expands so rapidly that it quickly encompasses the entire graph. It is likely that the real situation lies somewhere in between the extremes represented by the random graph model and the two local models that we have studied in this section and the previous one.
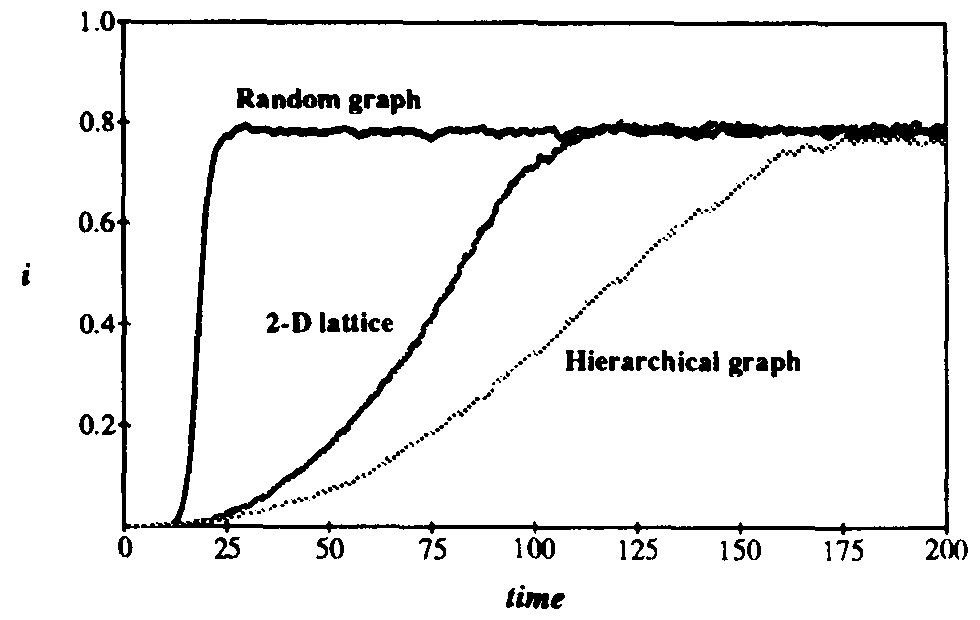
Figure 12: Comparison of fraction i of infected individuals vs. time for random graph, hierarchical graph, and two-dimensional graph SIS models. The infection and cure rates have their usual values of = 1.0 and = 0.2. The number of nodes in the simulations are 10000 for the random and spatial models and  = 8192 for the hierarchical model. In order to ensure that the equilibrium was close to the homogeneous limit, the connectivity of the random graph was chosen to be = 10, and the localization parameter for the hierarchical graph was r = 0.225. The parameters for the spatial graph are as given in Fig. 11. As predicted, the growth in the infected populationis quadratic in the spatial model, which is much slower than the exponential growth exhibited by the random graph model. The functional form of the growth of i in the hierarchical graph is not yet known.
= 8192 for the hierarchical model. In order to ensure that the equilibrium was close to the homogeneous limit, the connectivity of the random graph was chosen to be = 10, and the localization parameter for the hierarchical graph was r = 0.225. The parameters for the spatial graph are as given in Fig. 11. As predicted, the growth in the infected populationis quadratic in the spatial model, which is much slower than the exponential growth exhibited by the random graph model. The functional form of the growth of i in the hierarchical graph is not yet known.
Cohen showed that a perfect defense against computer viruses is impossible; we have shown that it may be unnecessary. Defense mechanisms are adequate for preventing widespread propagation of viruses if the rate at which they detect and remove viruses is sufficiently high relative to the rate at which viruses spread between users. The fact that an epidemic can only occur if the infection rate exceeds a finite critical threshold has been known in the biological realm for over half a century; we have shown that this result holds in the computational realm as well. For conditions which seem likely to hold in the computational domain, we have discovered that the epidemic threshold is actually higher than its classical value. Another encouraging finding is that, even if the infection rate is above the epidemic threshold, the number of infected individuals grows much more slowly than predicted by the standard homogeneous interaction model if the interactions are local.
In section 2, we formulated the directed random graph model and studied its behavior using three different techniques: deterministic approximation, stochastic approximation, and simulation. We obtained theoretical results which were essentially identical to those of the classical homogeneous interaction theory by ignoring the details of the connectivity of the graphs. In particular, we found that epidemics can not occur unless the ratio of the total rate at which an infected individual attempts to infect other individuals exceeds the rate at which individuals become cured. If the infection rate exceeds this threshold, an epidemic is still not certain, but it becomes increasingly probable as the infection rate is increased further above the threshold. The average number of infected individuals in equilibrium increases with the infection rate. It can take on any value, from near zero when the infection rate is just above threshold to the size of the population when the cure rate (and hence the threshold) is zero.
Simulations showed that these theoretical results hold when the connectivity of the graphs is sufficiently large, but fail miserably when the connectivity is small. In particular, if the total infection rate is held fixed while the connectivity is decreased, there is a dramatic decrease in the probability of an epidemic and in the average number of infected individuals. In other words, when the connectivity is small, the epidemic threshold is greatly increased over the value predicted by our extension of the classical homogeneous interaction theory. To our knowledge, this is the first observation of an apparent interaction between two well-known threshold phenomena: the epidemic threshold discovered by Kermack and McKendrick in the 1930’s [27] and the percolation threshold for random graphs discovered by Erdos and Rényi in 1960 [36].
In section 2.4, we added weak links to the random graph model to simulate the effect of infrequent program sharing with many other individuals. In this case, the epidemic threshold is intermediate between the random-graph and the homogeneous values. The hierarchical model of section 3 was introduced to account for a distribution of rates of program sharing in a more realistic manner than the weak-link model. In addition, it allowed us to capture the phenomenon of user cliques - groups of users which share programs with one another more frequently than with other users. We observed an increase of the epidemic threshold over its classical value which was very similar in character to that of the random graph model. Individual simulations of epidemics on hierarchical graphs revealed a number of interesting and surprising features. In some cases, the number of infected individuals fluctuated wildly; in others, the number of infections formed a series of plateaus separated by rapid growth spurts.
Taken together, the results of the random graph, weak-link, and hierarchical models demonstrate that, when most of the total infection rate is concentrated into just a few nodes, epidemics have a much harder time establishing themselves than predicted by the classical homogeneous theory. We are currently trying to develop theories which can describe this very interesting effect quantitatively.
By studying a spatial model in section 4, we viewed the effect of locality and user cliques from another perspective and obtained some analytic results based upon a diffusion-like equation for viral spreading in space and time. We found that the number of infected individuals grows polynomially in time, as opposed to the exponential growth rate in random graphs. The growth rate in the hierarchical model also appears to be polynomial under some conditions, but we have not yet obtained analytic results in this case. We believe that actual systems are intermediate between the extremes of random connectivity and local connectivity, so we expect the growth rate of infection to be intermediate between that of the random graph model and that of the hierarchical and spatial models.
The epidemiological approach to the study of computer virus propagation is quite general because it makes no assumptions about how viruses are detected and removed. Any mechanism that diminishes the infection rate or increases the detection rate will help to prevent widespread epidemics. The existence of a sharp threshold for epidemics means that it is worth doing everything possible to bring the infection rate below this threshold, but that further effort is not warranted. Our discovery that the topology of program sharing can have a profound effect upon the ability of viruses to spread may eventually lead to alternative methods for suppressing epidemics which could supplement the above-mentioned efforts to affect the infection and cure rates. While the models that we have studied are still somewhat simplistic, we expect that future work on more complex and realistic models will retain many of the features that we have observed here.
The work that we have presented here immediately suggests a number of areas for further research. We are currently trying to gain a better understanding of how the epidemic threshold depends upon the connectivity of a random graph. Our present understanding of the hierarchical model is solely based upon simulations, and we would like to develop theoretical expressions for the epidemic threshold as a function of the localization parameter and for the functional form of the growth rate of an epidemic. The peculiar phenomena that we have observed in individual simulations on hierarchical graphs, such as wild fluctuations and plateaus in the number of infected individuals as a function of time, deserve further attention. We would also like to experiment with disordered hierarchical graphs, in which the hierarchy of rates is retained but the locality of interactions is strongly disturbed or destroyed. This might allow us to isolate the effects of locality more cleanly.
We have not touched upon several areas that merit future investigation. A number of other models in the epidemiological literature have important analogs in the computational realm. In particular, the SIR (susceptible → infected → removed) model, in which individuals become permanently immune once they have been infected and cured, would be appropriate in the limit where users become extremely vigilant after having experienced a viral infection. The actual situation is probably somewhere between the SIS and SIR extremes. After discovering a viral infection, users may initially become much more conscientious about using anti-virus software, but if a long time passes without incident they may relax their vigilance to some degree. Models analogous to this scenario have been studied within a biological context, for there are some cases in which the body gradually loses its immunity to a particular disease [27]. Another interesting notion is the "kill signal", a message sent by a node upon discovering that it is infected, warning all nodes to which it is connected that they may also be infected. Our preliminary investigations suggest that this may be one of the most powerful means for thwarting epidemics. Certainly, it makes a good deal of intuitive sense and has long been used in the medical profession for the purpose of stamping out sexually transmitted diseases. The kill signal is just one of many examples of adaptive responses to viral infection. A close study of the immune system might prove to be a rich source of ideas for other adaptive methods for control and suppression of computer virus infections.
Finally, we feel that it is of the utmost importance to collect data on program-sharing habits and viral spread rates and incorporate them into our models. User surveys and centralised reporting of virus incidents would be invaluable. As epidemiologists have discovered, the task of collecting such information and incorporating it into models is fraught with difficulties, but we hope to benefit from the decades of experience that they have accumulated in dealing with such problems.
We are not the first to apply the mathematical techniques of epidemiology outside of the biological realm. Mathematical epidemiology has been used to gain insights into how ideas propagate [37] and, more practically, to develop novel algorithms for maintaining replicated databases [38]. As often occurs when mathematical techniques are adapted to new applications, we have been forced to extend those techniques in somewhat unfamiliar and unanticipated directions. We hope that in so doing we have enriched mathematical epidemiology and all fields to which it can be applied as much as we have benefitted from using it.
We are grateful to William C. Arnold, David M. Chess and Steve H. Weingart for useful conversations on how computer viruses behave in the real world, and to Scott Kirkpatrick for encouraging our interest in analytic and modelling approaches to this problem. We thank Paula K. Sweeney, David W. Levine and Paul J. Fink, Jr. for their assistance in harnessing the power of several IBM RISC System 6000 computers. In combination with a few IBM 3090s, the resultant 300 MIPS performance enabled our simulations to be completed in a few weeks.
1 The latter case is sometimes referred to as a “worm”, but since we are solely concerned with the property of self-replication, we shall call all such entities “viruses”.
2 We use the term “epidemic” to refer to any widespread, persistent infection in a population, even in cases where the fraction of infected individuals is extremely low.
3 The term “connectivity” is used in a differentsense by graph theorists.
4 These values for the infection and cure rates will be adhered to throughout this work to facilitate comparison of the various models. Since 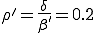 ,the infection rate is five times the classical homogeneous threshold of
,the infection rate is five times the classical homogeneous threshold of 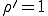 for epidemics that was derived in section 2.1.
for epidemics that was derived in section 2.1.
5 The event-drivensimulation has computational advantages over simulations which employ fixed time steps. Within a fixed time interval, no events can occur (which is inefficient), or several can occur, causing confusion about the order of the events within that interval. The event-driven simulation guarantees that exactly one event occurs per time step.
6 We use the term "clique" in its colloquial sense, not in its graph-theoretical sense.
[Back to index] [Comments (0)]