
Paul-Michael Agapow
Complexity International (1996) 3
1996
Traditionally, computer machine languages have been regarded as inappropriate for evolutionary activity. This is due largely to the perception that machine language is computationally brittle (that is, working programs are a small fraction of the total of all possible programs and therefore a random change to a functional program will almost certainly render it non-functional). This assumes, however, that legitimate program sequences are dissimilar to each other and that "brittleness" does not exist in biological evolutionary systems. The first assumption was tested by checking for homology between legitimite program sequences at the machine level. A surprising degree of similarity was discovered, vastly above what would be expected if legitimate code sequences were only randomly related. The second assumption was examined via available data on protein mutations and composition. Although robust in some aspects, terrestrial biology also demonstrates significant brittleness. The difference in brittleness between computational and terrestrial systems therefore ceases to be a qualitative gap and becomes quantitative, albeit perhaps a large one. In light of the findings, some intriguing possibilities are examined. The evolution "in the wild" of computer viruses is considered and it is concluded that while evolution de novo is still unlikely, viable mutation of one virus strain to new species is possible. This explains a number of mutant viruses that have been discovered. Also considered is evolutionary computation using machine language as a possible optimisation tool, as is the significance of brittleness in evolutionary systems.
The conventional von Neumann computer architecture has been the dominant paradigm for computation basically since its invention half a century ago. This is not without good reason; although there other feasible designs for computers, none has yet matched the von Neumann design's practicality and general power. However, there have been serious complaints levelled against it - charges that it is the very antithesis of more biology-influenced modes of computation like evolutionary computation and artificial life. (For example see [2].)
Prominent amongst the charges made is that the von Neumann architecture and programs are computationally brittle [19], [8]. That is to say, that the ratio of viable programs to the total number of possible programs is minuscule, and thus any random mutant of a functional program is almost certain to be nonfunctional. (This might be visualised in genetic terms as a sparse landscape of precipitous fitness peaks separated by wide gulfs of unfitness and dysfunction.) It follows, therefore, that evolution is unlikely. Belief in the truth of this thesis has shaped the design of evolutionary computational systems in an attempt to create models divorced from a machine architecture and its perceived unsuitability.
There are grounds, however, for doubting the validity of computational brittleness. The following work shall examine the above premise in a two-pronged attack. Firstly, the brittleness of conventional machine code sequences shall be examined in a series of homology studies. Secondly, the fragility in terrestrial biological systems shall be examined. These findings shall be used to cast a new and intriguing light on the possible evolution of computer viruses "in the wild" and aspects of evolutionary computation.
It is true that sophisticated behaviour has been observed in computational ecosystems based on a von Neumann paradigm. (For example [18], [5], [20], [19] and many others). One recent study [17] has even demonstrated the ability of such systems to self-organise replicating structures from random "inanimate" code. While it is tempting to hold these as evidence for the "non-brittleness" of machine code, this is not a valid comparison. All of these systems have made significant alterations to the von Neumann design that foster life-like processes, including greatly simplified machine languages that have been reinforced against mutation. Thus, these systems at the moment can tell us nothing about the significance of brittleness.
Considering conventional computation, brittleness as posed, assumes that functional programs are unrelated to each other. Put another way, in "program space" (the space of all possible programs) legitimate programs are distributed in a largely random manner. However, it seems intuitively obvious that functional programs should have similar sequences to other functional programs. This is borne out by the not infrequent misidentification of legitimate software as viral by anti-viral software scanning for virus sequences [21].
In order to test homology in functional real world program sequences, a series of executable binaries on a run-of-the-mill Unix computer (a SPARCstation running SunOS 4.1) were converted into hexadecimal code. (Note, that thus each byte was translated into 2 hexadecimal numbers.) The executables were selected for their ubiquity and representation of a variety of functions. Those that obviously shared common functions or purpose, or those containing a great deal of ASCII string data were not used.
The software used for matching of homologous sequences was a modified version of the Unix tool agrep (approximate grep [24]), which can search text and report the number of matches for a sequence within a given limit of substitutions, insertions and deletions. The choice of this software above more specialised DNA/protein sequence searchers was made for the ease of preparing the data, the relatively minor modifications necessary to the search engine and the ability to weight certain types of matches. It should be noted that this approach, matching on the half-byte ("nybble") level, will actually under-estimate homology. For example, the hex values 9 and 8 will be seen as a complete mismatch despite the fact that they differ by only 1 bit.
Sequences of various lengths were randomly extracted from each executable's hexdumps and matches of varying degrees of homology searched for in the other hexdumps. The pattern of homology was complex but generally high, with virtually all short (less than 64 bytes) sequences finding multiple matches within a moderate amount of stringency, with some finding literally thousands of matches. This decreased with longer sequences (which computational demands limited the longest sequence to 128 bytes) but not as sharply as might be expected (see Figure 1). Quite surprising was the large number of sequences that were perfect matches (that is, zero substitutions, deletions or insertions were required to match) even at long sequence lengths.
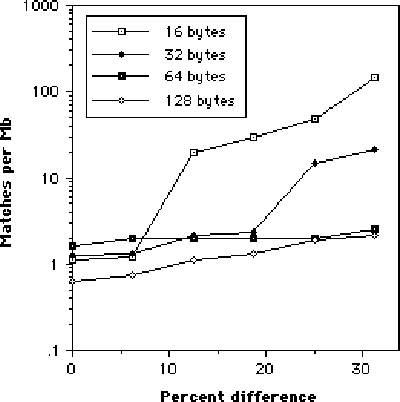
Figure 1: Sequence homology. Each plot represents the mean success rate for 15 search sequences over 4 megabytes of data. A match of x% means that the target sequence differed by only x% or less from the search sequence.
Further searches were made for matches allowing only substitutions (effectively point mutations). However this resulted in only a slight lowering of homology (see Figure 2). It is also possible that the homology seen might be the result of code from libraries included at program compilation. Testing this hypothesis is more difficult due to the lack of nontrivial programs that do not use common libraries. Nonetheless, a small number (five) were obtained and treated as above. Again there was only a small lowering in homology, although due to the small number of programs used, this result should be treated with caution.
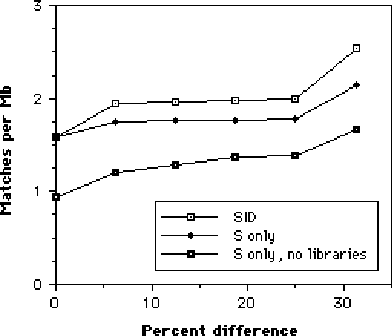
Figure 2: Homology with substitution only. 64-byte sequences matched for homology as previously. "SID" indicates the previous plot, with substitutions, deletions and insertions allowed. "S only" indicates only substitutions were allowed. "No libraries" indicates matches over programs that did not include library code.
The level to which the homology seen is above background (that is, simple coincidence) may be assessed thus: a 64-byte sequence is 512 bits. Therefore it will perfectly match other randomly generated 64-byte sequences 1 in every 2512 trials. From the homology data above (using the search with substitution only and without included libraries) a perfect match is found about once every megabyte. As matching is done on the nybble level, in every megabyte there are nearly 221 sequences of 64 bytes that can serve as possible matches. As 64-byte sequences have, on average, a perfect match in every megabyte, they therefore show a homology of 1 in 221, or 2491 times more than expected.
Therefore, in summary, functional program sequences are much more likely to be similar than previously thought. In fact, legitimate sequences in "program space" would seem to be very tightly clustered in some cases. The chance of a functional code sequence mutating to another appears quite possible.
The positioning of computational brittleness as a barrier to evolution in machine language implies that such a barrier does not exist in terrestrial genetic systems. Put another way, this supposes that mutation in biological systems generally does not end in dysfunction. This allegation bears examination.
For reasons of brevity, readers are referred to any molecular biology textbook for full details on the terrestrial genetic code. To recap its salient features: genes (in the form of DNA) are transcribed into RNA which is then used to produce a polypeptide. The code is based on triplets of nucleotides or codons, where each nucleotide is 1 of 4 possibilities (A, C, G or U) and sequential codons indicate sequential amino acids on a protein chain. (By convention, the code is referred to here as it holds for RNA, although a complementary code exists, of course, for the DNA from which it is transcribed.) A degeneracy exists in the code such that a particular amino acid is produced by more than one codon. There are some special codons - AUG (methionine, an initiation signal that is the first codon of nascent RNA strands) and UAG, UAA, UGA ("stop" codons which do not stand for any amino acid but for termination of polypeptide synthesis). Note, there is no "punctuation" in the code; there is no separation between adjacent codons. Finally, some editing and processing does occur at points during and after transcription and translation but this does not affect the analysis below.
Simple mutations that alter the DNA (and hence RNA) sequence, therefore, are of several types:
To judge the brittleness of the terrestrial genetic code it would be useful to find what proportion of mutations effect a transition between two functional genes. Unfortunately, the available data is clouded by a variety of factors. Many studies involve many different mutagens, each having characteristic and different types of mutations [1]. Further, the dosages used often lead to severe genetic lesions that are significantly more lethal than the simple mutations above. Also, many studies show a selectivity in the sites they have studied, a bias due to (for example) the mutagens used or simple experimental interest. Thus, there is perhaps a gap between these laboratory exercises, mutation in the wild and idealised assessments of brittleness. This is further complicated by the issue of the effects of mutations being at the heart of the neutralism debate [11], where estimates of how many substitutions result in significant functional change range from virtually 0 to 100% [10].
With these caveats, the classic experiments of Muller [16] and more recent work [7] indicate that the majority of detectable mutations are deleterious to their host. However, these studies involved fairly severe mutagenesis and, as such, are subject to the criticism above. Other studies have examined the effect of random mutations on protein-coding genes. Excluding those in which mutations may have been in some way selected, around 40% of missense mutations lead to dysfunction (see Table 1). (In this case, "dysfunction" means either no detectable protein was produced, that the protein produced had little detectable activity or the mutation was lethal.)
| Target Protein | % Dysfunctional | Reference |
|---|---|---|
| lac repressor | 42 | [15] |
| tryptophan synthetase A | 58 | [25] |
| tobacco mosaic virus | 25 | [23] |
| hemoglobin | 32 | [22] |
Table 1: Missense Mutations. Collected data for the percentage of proteins that became dysfunctional upon a single amino acid substitution.
Some data can be obtained by examining what mutations persist in terrestrial life. It can be shown that the actual substitution frequency distribution (that is, the relative frequencies of the various missense mutations) is vastly different from the theoretical distribution. In fact, a large number of possible substitutions - more than 50% - were not observed at all in a sample size of more than 1500 proteins [6]. It is generally and reasonably assumed that this discrepancy is explained by the missing substitutions being those which disrupt the protein function as such mutations will be quickly lost [4]. Note, that this is certainly an underestimate as the 50% includes only those substitutions that never occur.
Finally, an estimate of the proportion of substitutions that result in dysfunction can be obtained by looking at the ratio of samesense (silent) mutations to missense mutations. A list of mutations at 43 mammalian loci [14] shows a ratio of substituting mutations to silent mutations through a range of 0 - 0.6 to 1. (The average is around 0.25 to 1.) The ratio of nucleotide changes that lead to missense as to samesense is 393 to 156 (see below) or 2.54 to 1. Reasoning as above that the missing substitutions are those leading to dysfunction, around 90% of missense mutations appear to sufficiently deleterious as to be selected into non-existence. (By similar reasoning, Lewin [13] has also arrived at 90% for the beta and delta globin genes.)
Given the limited amount of data involved, caution in analysis is advised. Most of the above calculations assume that dysfunctional mutations are quickly lost, substitutions are independent of each other and that amino acids are used in roughly equal amounts. Each of the assumptions is false to some degree but allowing for their effect is problematic. Nonetheless, it is seems clear that in the vicinity of 50% of missense mutations will lead to dysfunction. (Note, that substitution is certainly less constrained in structural proteins, as opposed to the functional ones listed above. For purposes of comparison to computer programs however, functional mutations are a more appropriate model.)
To give an impression of the above figures, imagine mutations in an ideal protein 100 amino acids long - short compared to some eukaryotic proteins, but not unreasonably so and close to the length of the ubiquitous cytochrome c family. The DNA coding region for the protein is therefore 303 bases long - 3 bases for amino acid codon and the termination codon. It is assumed that nonsense mutation or any change to the initiation codon will result in functional inactivation and that the coding region otherwise uses each codon equally; that is, the 61 amino acid codons are used evenly amongst the remaining 99 codons of the gene, and therefore each codon is used (99 / 61) 1.62 times. Each base can mutate to one of the three others and so there are 909 immediate "mutation neighbours" of the coding region. The initiation codon may mutate in 9 different ways and the three possible termination codons may mutate to 8, 8 and 9 non-termination (that is, functionally different) codons. Of the 61 amino acid codons, 19 may mutate to a termination codon. Therefore of the 909 possible mutations available, at least 9 + (8 + 8 + 9) / 3 + (19 * 1.62) = 48.1 or 48.1 / 909 = 5.3% will result in inactivation of the product via initiation or nonsense mutations.
The number of codon changes that resulted in missense mutations (excluding the stop codons) was counted as 393 from a total of (9 * 61) 549, or 71.6%. If we assume (as above) that roughly 50% of these missense mutations will result in dysfunction and allow for the catastrophic mutation of the initiation codon or of other codons to nonsense codons as above, we arrive at a figure of 40.4% point mutations resulting in dysfunction.
Note, that if the occurrence of the generally catastrophic frameshift (insertion / deletion) mutations is included, these figures will actually rise. In summary therefore, it seems that while there is some degeneracy in the terrestrial genetic code, there is also a great degree of brittleness. In fact, it is likely there is more brittleness than there is degeneracy.
It is therefore clear that machine language is considerably less brittle and the terrestrial genetic code more brittle than might previously have been expected. It is difficult to arrive at an appropriate measure for comparing the two, but the gap between them would seem to be no longer qualitative but quantitative, albeit perhaps still a large one. A number of striking ideas follow from this.
Firstly, computational brittleness has been seen as a barrier to the evolution of computer viruses. Calculations have been produced that render the probability of a virus arising from non-viral code as vastly improbable, the time involved being "several times the lifespan of the universe" [9], [3]. Yet given the homology seen between program sequences here (up to 128 bytes, on the lower but feasible end of computer virus functionality) this probability should be reassessed. Burger's calculation [3], based on a 1000-bit long virus yields a probability of 1.22 * 10-366. If this calculation is made instead, allowing for a more (primordial) virus and greater homology, we find that the probability of evolution for a 500-bit, 60% homologous sequence is 1.27 * 10-165. If we assume 80% homology, the result is 1.18 * 10-112. These are still extremely poor odds, but coupled with an appreciation of brittleness, explains the number of spontaneously mutated computer viruses seen in the wild [9].
Next, if machine language is considerably less brittle than imagined, this opens up the idea of using machine code as an arena for evolutionary computation. Such a low-level and platform-specific representation is unlikely to supplant other evolutionary computation paradigms, but does offer two intriguing advantages. A machine code capable of universal computation does not suffer from the symbol sufficiency problem, whereas the alphabet for an evolutionary computation must be equipped with adequate words to describe a solution [12]. Also, a machine-level representation may prove to be a powerful device in microcode optimisation.
Finally, we are led to reflect upon the role of brittleness in evolutionary systems. The fact that brittleness exists substantially in biology is perhaps a clue that it (or some underlying feature of which it is the unavoidable consequence) fulfils some important function. As a simple example, brittleness could be the result of restricting the connectivity of different genotypes, allowing a genotype to directly evolve to only a constrained set of others and thus avoid premature convergence and loss of diversity within a population. Whatever the underlying reason, elucidation is likely to have a significant bearing on population genetics and the issue of representation in evolutionary computation.